Síndrome de Muir-Torre
| Síndrome de Muir-Torre | ||
|---|---|---|
 Fotografía de un sebaceoma o epitelioma sebáceo cutáneo que hace sospechar del síndrome de Muir-Torre. | ||
| Especialidad | Oncología, dermatología y genética médica | |
| Síntomas | Adenocarcinoma o adenoma sebáceo y/o epitelioma faciales | |
| Inicio habitual | Más de 50 años | |
| Causas | Mutación genética heredable | |
| Tratamiento | Quirúrgico (tumorectomía y colectomía) | |
| Medicación | Isotretinoína | |
| eMedicine | derm/275 | |
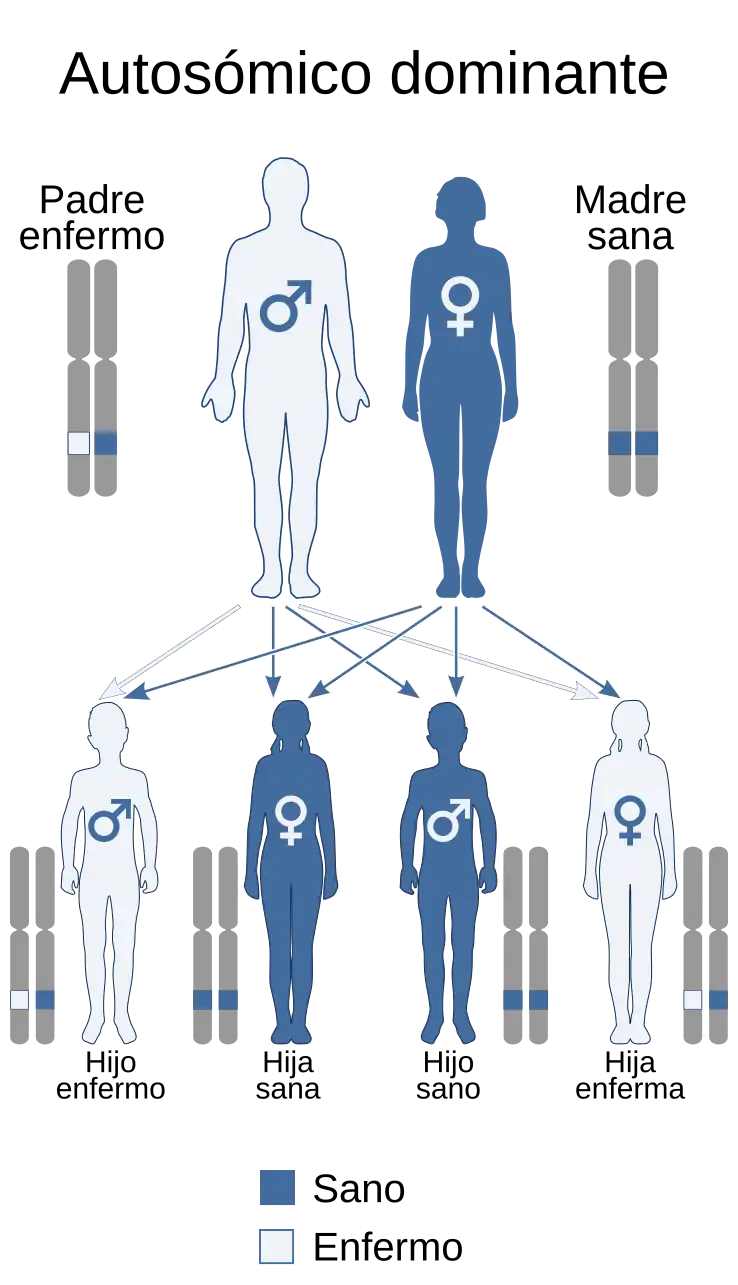
El síndrome de Muir-Torre es una genodermatosis o bien una enfermedad hereditaria autosómica dominante por la mutación de los genes MLH1 y MSH2 principalmente pero también puede estar afectado el MSH6, que combina por lo menos una de las neoplasias cutáneas —ya sea adenoma o carcinoma sebáceos en primer lugar pero también podría ser un sebaceoma o epitelioma sebáceo, con o sin queratoacantoma— y el cáncer colorrectal en la mayoría de los casos (en el 61%) que se suele manifestar después de los 50 años, con una incidencia del doble en hombres que en mujeres y suelen ser de bajo grado, inicio precoz y permiten una prolongada supervivencia. En un muy bajo porcentaje se podría combinar con el síndrome de Turcot y se considera una variante fenotípica del síndrome de Lynch.
Causas genéticas

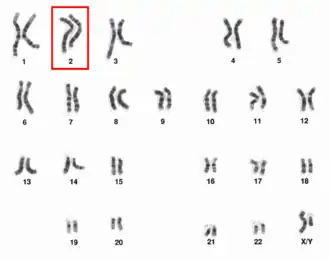
Las causas del síndrome de Muir-Torre son las mutaciones de los genes MSH2 —que es el más frecuentemente mutado que se encuentra en la brazo corto del cromosoma 2 submetacéntrico o bien cromosoma 2p— y con menor frecuencia el MLH1 —situado en el cromosoma 3— y que están implicados en el 90% de las familias afectadas por este síndrome, aunque también puede estar mutado el gen MSH6 en alrededor del 7-10% de los casos (por lo que podría aparecer simultáneamente con el síndrome de Turcot). Dichas mutaciones se transmiten de padres a hijos según un patrón autosómico dominante,[1] de alto grado de penetrancia y con una expresividad variable.[2]
Los genes que están implicados en el mecanismo de reparación de desapareamientos de ADN son siete: MSH2, MLH1, MSH6, MLH3, MSH3, PMS1 y PMS2, y las siete proteínas codificadas por los mismos, forman un complejo que actúa al final de la «fase S» del ciclo celular y reconocen los apareamientos erróneos de bases nitrogenadas en el ADN, causados por errores de replicación, por lo que activan a la exonucleasa I, las proteínas SSB y la helicasa para que consecuentemente puedan actuar la ADN polimerasa y ligasa para repararlos.[1]
Dichos errores de replicación de ADN ocurren con mayor frecuencia en microsatélites debido a su naturaleza repetitiva y cuando falla la maquinaria de reparación post-replicativo y el cambio permanece, puede producirse la inestabilidad en los mismos y esta puede llevar a la acumulación de mutaciones somáticas a lo largo de la vida del individuo.
Sintomatología
Los síntomas del síndrome de Muir-Torre, que se considera una variante del síndrome de Lynch,[3] corresponden al menos por una neoplasia cutánea sumado a por lo menos un cáncer visceral, la proporción hombre/mujer es 2 a 1 y la media de edad se sitúa alrededor de la quinta o sexta décadas de vida:[2]
- Lesiones dermatológicas: aparición al menos de un adenoma, adenocarcinoma sebáceo o epitelioma y podría aparecer o no un queratoacantoma o varios. La sola presencia de una neoplasia cutánea con la mutación genética no significa que se padezca el síndrome, por eso es necesario confirmar el siguiente síntoma que en la mayoría de los casos aparece anteriormente.[2]
- Cánceres viscerales: manifestación de al menos un cáncer interno ya sea del tracto digestivo como el colon y el recto (en el 61% de los casos con cáncer colorrectal) o urogenitales (en el 22% de los casos).[1] Otros tipos de cáncer pueden afectar el intestino delgado (en el 4 % de los casos), los ovarios, el útero y las mamas (también en bajos porcentajes). Suelen ser de bajo grado, inicio precoz y permiten una prolongada supervivencia.[2]
Criterios de Ámsterdam
Los criterios de Ámsterdam sirven para identificar a las personas candidatas a realizarse un examen genético que demuestre la existencia del síndrome:[4]
- Tres o más familiares con un cáncer asociado a HNPCC (es decir, cáncer colorrectal sin poliposis, cáncer de endometrio, intestino delgado, uréter o pelvis renal), uno de ellos debe ser pariente de primer grado de los otros dos.
- Dos o más generaciones sucesivas afectadas por cáncer.
- Uno o más personas de la familia con cáncer de primer grado de las otras 2, al menos 1 caso de cáncer colorrectal diagnosticado antes de los 50 años.
- Los tumores se verifican mediante examen histológico pero debe excluirse el diagnóstico de poliposis adenomatosa familiar.
Diagnóstico
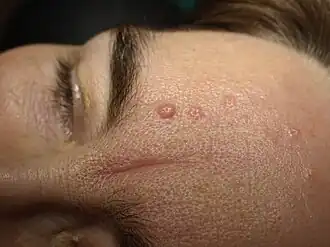
En la exploración dermatológica de pacientes de más de 50 años de edad, si hubiera alguna presencia de un área facial de una o múltiples lesiones papulosas, rosadas, color carne o amarillentas, con hoyuelo central y un diámetro inferior a 1 cm, hay que remitir al examen histopatológico para confirmar diagnóstico mediante técnica de inmunohistoquímica de adenomas o adenocarcinomas.[1] Las dichas neoplasias cutáneas aparecen posteriormente a los carcinomas viscerales en el 56% de los casos, también pueden precederlos en un 22% e inclusive aparecer simultáneamente en un 6% de los casos.[2]
También se recomienda realizar pruebas complementarias como una analítica general, marcadores tumorales, hormonas tiroideas, un TAC toracoabdominal, colonoscopia y endoscopia digestiva alta. Con las anteriores exploraciones dermatológicas y complementarias se llega a un diagnóstico diferencial ya que hay que descartar verrugas por VPH y molusco contagioso por poxvirus, además por la sola presencia de nevus o hiperplasias sebáceas se descartaría el síndrome.[2]
Un estudio italiano del 2015 sugirió que la activación del sistema de las glándulas sebáceas, incluidas las hiperplasias sebáceas intraorales o manchas de Fordyce de la mucosa oral que ocurre en pacientes con ETS, puede constituir un parámetro clínico adicional del síndrome de Muir-Torre, y podría distinguir a los individuos con mayor probabilidad de ser afectados por este síndrome.[5]
Tratamiento
El tratamiento del síndrome de Muir-Torre consiste en la administración de isotretinoína por vía oral. Se ha demostrado que este fármaco previene el desarrollo de tumores cutáneos y viscerales, además de hiperplasias sebáceas. Para los carcinomas intestinales el tratamiento es quirúrgico mediante hemicolectomía.[2]
Los pacientes sospechosos de padecer este síndrome deben someterse a las mismas pruebas rigurosas de detección del carcinoma colorrectal y otras neoplasias malignas, al igual que los pacientes con síndrome de Lynch. Esto incluye colonoscopias frecuentes y tempranas, mamografías, evaluación dermatológica y estudios de imagen del abdomen y la pelvis.[2]
Véase también
- Desorden de deficiencia en la reparación del ADN
- Manchas de Fordyce
- Poliposis adenomatosa familiar
- Síndrome de Lynch
- Síndrome de Turcot
Referencias
- ↑ a b c d Grandhi, 2013, p. 52.
- ↑ a b c d e f g h Gómez-Centeno, 2003, pp. 42-44.
- ↑ Roberts, 2014, pp. 711-716.
- ↑ Vasen, 1999, pp. 1453-1456.
- ↑ Ponti, 2015, pp. 552-557.
Bibliografía
- Gómez-Centeno, Pilar, con Rodríguez López, José Ángel;García-Costa, Agustín; Álvarez López, José; Cabo Gómez, Fernando; Veiga Codesido, Carlos; Fonseca Moretón, Arístides, y Gómez Domínguez, José Miguel (2003). Actas Dermo-Sifiliográficas: Síndrome de Muir-Torre. 94 (1). Academia Española de Dermatología y Venereología. ISSN 0001-7310.
- Grandhi, R., con Deibert, C.P.;Pirris, S.M.; Lembersky, B., y Mintz, A.H. (2013). Surgical Neurology International: Simultaneous Muir–Torre and Turcot's syndrome: A case report and review of the literature. 4 (52). ScientificScholar. PMID 23646262.
- Ponti, G., con Meschieri, A.; Pollio, A.;Ruini, C.; Manfredini, M.; Longo, C., y Pellacani, G. (2015). Fordyce granules and hyperplastic mucosal sebaceous glands as distinctive stigmata in Muir–Torre syndrome patients: characterization with reflectance confocal microscopy. 44 (7). Journal of Oral Pathology & Medicine.
- Roberts, M.E., con Riegert - Johnson, Douglas L.; Brittany, C. Thomas; Leppig, Kathleen A., y Lim, Justin (2014). Genetis in Medicine: A clinical scoring system to identify patients with sebaceous neoplasms at risk for the Muir–Torre variant of Lynch syndrome. 16 (9).
- Vasen, Hans F.A., con Watson, P.;Mecklin, J.P.; Lynch, H.T. (1999). Gastroenterology: New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. 116 (6). Academia Española de Dermatología y Venereología. PMID 10348829.
Enlaces externos
- Síndrome de Muir-Torre
- Criterios de Ámsterdam
- Gránulos de Fordyce y glándulas sebáceas mucosas hiperplásicas como estigmas distintivos en pacientes con síndrome de Muir-Torre: caracterización con microscopía confocal de reflectancia
- Un sistema de puntuación clínica para identificar a pacientes con neoplasias sebáceas en riesgo de padecer la variante Muir-Torre del síndrome de Lynch
- Síndrome de Muir-Torre y Turcot simultáneos: reporte de un caso y revisión de la literatura
- En RARe SOURCE
- Artículos en PubMed