Ácido trifluoroperacético
| Ácido trifluoroperacético | ||
|---|---|---|
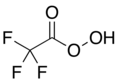 | ||
| Nombre IUPAC | ||
| Ácido trifluoroetanoperoxoico | ||
| General | ||
| Otros nombres |
| |
| Fórmula estructural |
| |
| Fórmula molecular | ? | |
| Identificadores | ||
| ChemSpider | 8466281 | |
| PubChem | 10290812 | |
| UNII | PU9V6QCS3Q | |
| Propiedades físicas | ||
| Masa molar | 129,987779 g/mol | |
El ácido trifluoroperacético (ácido trifluoroperoxiacético, TFPAA) es un compuesto organofluorado, el análogo peroxiacético del ácido trifluoroacético, con la fórmula estructural condensada CF3COOOH. Es un agente oxidante fuerte para reacciones de oxidación orgánica, como en las oxidaciones de Baeyer-Villiger de cetonas.[1]Es el más reactivo de los peroxiácidos orgánicos, lo que le permite oxidar con éxito alquenos relativamente poco reactivos a epóxidos, donde otros peroxiácidos son ineficaces.[2]También puede oxidar los calcógenos en algunos grupos funcionales, como al transformar selenoéteres en selones.[3]Es un material potencialmente explosivo[4] y no está disponible comercialmente, pero se puede preparar rápidamente según sea necesario.[5]Su uso como reactivo de laboratorio fue iniciado y desarrollado por William D. Emmons.[6][7]
Propiedades
A temperatura y presión ambientales normales, el ácido trifluoroperacético es un líquido incoloro con un punto de ebullición de 162 °C.[8]Es soluble en acetonitrilo, diclorometano, éter dietílico y sulfolano, y reacciona fácilmente con el agua.[5]Al igual que todos los peroxiacidos, es potencialmente explosivo y requiere un manejo cuidadoso.[4]No se comercializa, pero se puede fabricar en el laboratorio y almacenar hasta varias semanas a −20 °C.[5] Algunos métodos de preparación dan lugar a mezclas que contienen peróxido de hidrógeno residual y ácido trifluoroacético, y calentar dicha mezcla es extremadamente peligroso; el peróxido de hidrógeno se puede descomponer utilizando dióxido de manganeso por seguridad antes de calentar.[5][8]
Preparación
El ácido trifluoroperacético se puede preparar fácilmente mediante un proceso de síntesis orgánica[9]que consiste en tratar el anhídrido trifluoroacético con una solución acuosa concentrada (90 %)[2]de peróxido de hidrógeno:
CF
3COOCOCF
3 + H
2O
2 → CF
3COOOH + CF
3COOH
Dado que el anhídrido forma ácido trifluoroacético al entrar en contacto con el agua, un exceso de anhídrido también sirve para eliminar el disolvente del reactivo peróxido:[9]
CF
3COOCOCF
3 + H
2O → 2 CF
3COOH
Se puede utilizar una solución de peróxido de hidrógeno más diluida (30 %) para formar ácido trifluoroperacético para algunas reacciones a partir del ácido trifluoroacético.[2]
CF
3COOH + H
2O
2 → CF
3COOOH + H
2O
Para evitar el peligro que supone manipular soluciones puras o muy concentradas de peróxido de hidrógeno, se puede utilizar peróxido de hidrógeno y urea para obtener el perácido.[5]Este método no utiliza agua, por lo que se obtiene un perácido completamente anhidro,[10] lo que supone una ventaja cuando la presencia de agua provoca reacciones secundarias durante determinadas reacciones de oxidación.[11]
CF
3COOCOCF
3 + H
2O
2·CO(NH
2)
2 → CF
3COOOH + CF
3COOH + CO(NH
2)
2
En los casos en los que se necesita un agente tampón del pH para una síntesis y se tolera la presencia de agua, se ha desarrollado otro método. Al hacer reaccionar el anhídrido trifluoroacético con percarbonato sódico, 2Na2CO3·3H2O2, se obtiene ácido trifluoroperacético y carbonato sódico, lo que evita la necesidad de un tampón adicional.[5][12]
3 CF
3COOCOCF
3 + 4 Na
2CO
3·32H
2O
2 → 6 CF
3COOOH + 4 Na
2CO
3 + 3 H
2O
El ácido trifluoroperoacético también se puede generar in situ,[13] lo que permite que reaccione rápidamente con el sustrato objetivo en lugar de sintetizar previamente un lote del reactivo para su uso posterior.
Historia y usos
iodo)benzene-3D-balls.png)
6H
5I(OOCCF
3)
2
El ácido trifluoropropanoico se utiliza principalmente como agente oxidante.[5][7] En septiembre de 1953, la revista Journal of the American Chemical Society publicó un trabajo de William D. Emmons y Arthur F. Ferris en el que se informaba de que este reactivo, generado in situ, era capaz de oxidar la anilina a nitrobenceno.[13]Durante los dos años siguientes, Emmons describió un método de preparación de este reactivo y publicó otros seis artículos en esta revista sobre sus aplicaciones.[14][15][16]Emmons es recordado en parte como el pionero[6] y desarrollador[7]del ácido trifluoroperacético como reactivo de laboratorio, que desde entonces se ha convertido en un reactivo útil para muchos tipos diferentes de reacciones sintéticas.
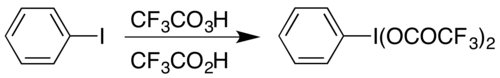
Un ejemplo es la formación del compuesto de yodo hipervalente (bis(trifluoroacetoxi)yodo)benceno, (CF3COO)2IC6H5 que se utiliza para llevar a cabo la reordenación de Hofmann en condiciones ácidas.[17] El compuesto hipervalente se puede obtener de dos maneras, y la elección suele depender de los materiales disponibles: se puede preparar a partir de su análogo acetato mediante una reacción de intercambio,[18]o haciendo reaccionar yodobenceno con una combinación de ácido trifluoroperacético y ácido trifluoroacético:[17]
Oxidación de Baeyer-Villiger
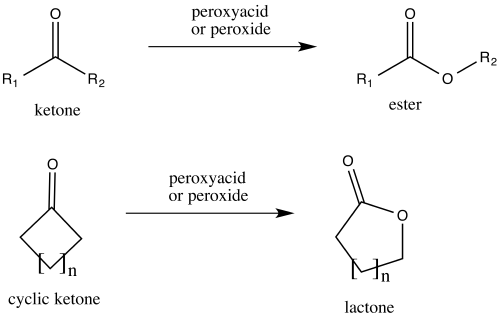
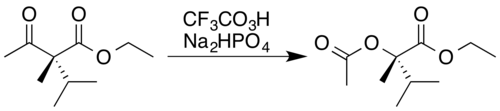
El ácido trifluoroperacético es uno de los reactivos más potentes utilizados para las oxidaciones de Baeyer-Villiger, debido a su alta acidez en comparación con otros perácidos y peróxidos similares:[19] 17 Esta reacción convierte las cetonas en ésteres de cadena lineal o lactonas, y recibe su nombre de Adolf von Baeyer y Victor Villiger, quienes la describieron por primera vez en 1899.[1]Se cree que la reacción se produce a través de un intermediario de Criegee[5] y muestra una buena regioselectividad y quimioselectividad para la posición de inserción del átomo de oxígeno, junto con la retención de la estereoquímica en la posición adyacente, como se puede ver en el siguiente ejemplo. El fosfato disódico (Na2HPO4) se añade como tampón de pH[2] para evitar que el subproducto trifluoroacético, altamente ácido, provoque la hidrólisis[20]o la transesterificación[21] del producto éster.
Epoxidación
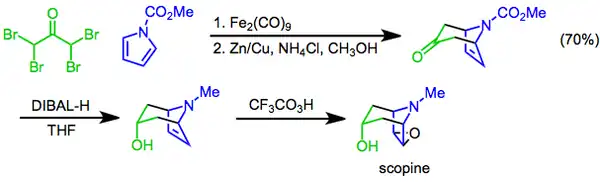
La reacción de Prilezhaev consiste en la conversión de un alqueno en un epóxido utilizando un perácido como oxidante[22] y se describió por primera vez en 1909.[23]La reacción se ha utilizado como paso final en la síntesis de la escopina, un alcaloide tropánico. En este enfoque, se utiliza una adición cíclica [4+3] mediada por diiron nonacarbonilo para construir el esqueleto bicíclico, a continuación se introduce el grupo funcional hidroxilo mediante la reducción diastereoselectiva de la cetona con hidruro de diisobutilaluminio, y la preparación se completa con una epoxidación con ácido trifluoroperacético de Prilezhaev.[24]
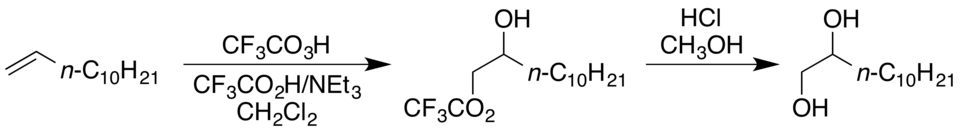
La alta reactividad del ácido trifluoroperacético en comparación con otros ácidos peroxi permite oxidar con éxito alquenos relativamente pobres en electrones, como el 1-hexeno, y ésteres α,β-insaturados, como el metacrilato de metilo, sustratos que suelen ser resistentes a la epoxidación con ácidos peroxi.[2] La inclusión de ácido trifluoroacético tamponado adicional en la mezcla da lugar a una estructura hidroxi-trifluoroacetato vicinal en lugar de un epóxido, que puede convertirse en diol mediante tratamiento con metanol ácido, como en la siguiente conversión de 1-dodeceno a 1,2-dodecanodiol.[2]
En el caso de un compuesto de alcohol alílico con un grupo funcional carbonilo próximo, el epóxido puede sufrir una reacción de expansión del anillo para formar un dioxolano.[5][11]El proceso que se describe a continuación se utilizó como parte de la síntesis total del neosporol, un producto natural:[11][25]
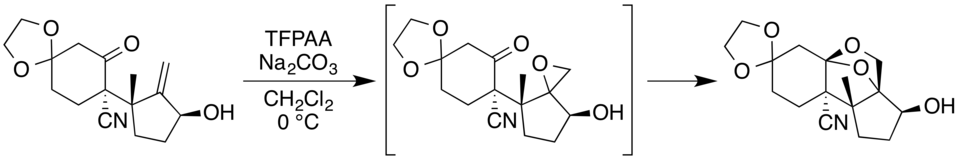
La preparación del compuesto isomérico esporol implicó una formación similar de dioxolano. En este caso, el uso de ácido trifluoroperacético derivado del peróxido de hidrógeno, que por lo tanto presumiblemente contenía trazas de agua, dio lugar principalmente a un hemiacetal en lugar del dioxolano de anillo cerrado. El uso del complejo de urea, que dio lugar a un material sin agua, permitió obtener con éxito el dioxolano como producto principal.[11]El dioxolano se expande al sistema 1,3-dioxano que se encuentra en el esporol en una etapa posterior de la síntesis.[25]
Oxidación de heteroátomos
Los grupos funcionales que contienen heteroátomos en estados de oxidación bajos pueden oxidarse con ácido trifluoroperacético.[5] [7]Entre los casos más comunes se encuentran la oxidación del yodo (por ejemplo, la formación del compuesto de yodo hipervalente a partir del yodobenceno mencionado anteriormente), el nitrógeno, el azufre y el selenio.
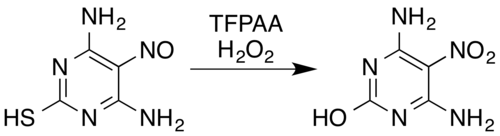
En el caso de los compuestos que contienen nitrógeno, las transformaciones conocidas incluyen oximas[5] y aminas primarias aromáticas[15] a compuestos nitro[7] (incluso con sustituyentes electronegativos, por ejemplo, pentafluoroanilina a pentafluoronitrobenceno),[26] nitrosaminas a nitraminas, [14][7] la formación de N-óxidos aromáticos y N-óxidos de azina aromáticos,[5][27] y la conversión de compuestos nitrosos en compuestos nitrosos o nitraminas. [5] Por ejemplo, una mezcla de peróxido de hidrógeno y ácido trifluoroperacético oxida la pirimidina sustituida con nitroso 4,6-diamino-5-nitrosopirimidina-2-tiol a su análogo nitro, al tiempo que elimina el grupo tiol mediante desulfuración hidrolítica oxidativa:[5][28]
En el caso de los elementos calcógenos, los grupos sulfuro (R–S–R) pueden oxidarse mediante ácido trifluoroperacético a formas de sulfóxido (R–S(O)–R) y/o sulfona (R–S(O)2–R), dependiendo de las condiciones utilizadas.[5] En el sistema análogo del selenio, la oxidación con ácido trifluoroperacético de los selenoéteres (R–Se–R) produce selones (R–Se(O)2–R) sin que se formen los selenóxidos relacionados (R–Se(O)–R) como producto aislable,[3] una reacción que es particularmente eficaz cuando R es un grupo arilo.[29] Un enfoque general para la formación de cloruros de sulfinilo (RS(O)Cl) es la reacción del tiol correspondiente con cloruro de sulfurilo (SO2Cl2). En los casos en los que se obtiene cloruro de sulfenílico (RSCl), una oxidación posterior con ácido trifluoroperacético proporciona el producto deseado, como en el caso del 2,2,2-trifluoro-1,1-difeniletanotiol:[30]
Cl.png)
La oxidación del tiofeno con ácido trifluoroperacético ilustra las vías de reacción competitivas, siendo posibles tanto la oxidación S como la epoxidación.[31] La vía principal forma inicialmente el sulfóxido, pero este compuesto químico sufre rápidamente una dimerización de tipo Diels-Alder antes de que se produzca una oxidación adicional; entre los productos de la reacción no se encuentran ni el tiofeno-S-óxido ni el tiofeno-S,S-dióxido.[5][31]El dímero puede oxidarse aún más, convirtiendo uno de los restos de óxido de azufre en dióxido de azufre. En la vía de reacción secundaria, una epoxidación de Prilezhaev[22]da lugar a la formación de epóxido de tiofeno-2,3, que se reorganiza rápidamente en el isómero tiofeno-2-ona.[32] Los experimentos de captura[31]demuestran que esta vía del epóxido no es una reacción alternativa del intermediario óxido de azufre, y los experimentos de marcado isotópico demuestran que se produce un desplazamiento 1,2-hidruro (un desplazamiento NIH) y, por lo tanto, que interviene un intermediario catiónico.[31] La elección del método de preparación del ácido trifluoroperacético es importante, ya que el agua suprime la vía de reacción secundaria, probablemente porque actúa como base competidora.[31]
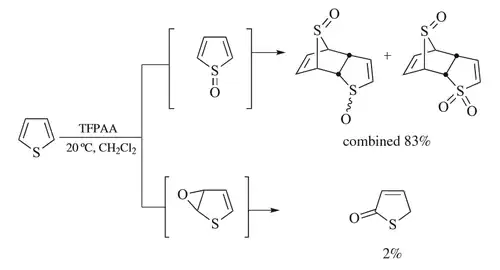
Oxidación con reordenamiento ácido
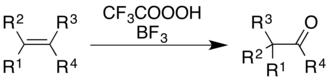
El uso de ácido trifluoroperacético con trifluoruro de boro provoca la oxidación de alquenos y anillos aromáticos con el consiguiente reordenamiento del esqueleto molecular.[5]
En el caso de los alquenos, la reacción da lugar a un producto cetónico, aunque el proceso mecánico no es simplemente una epoxidación seguida de un reordenamiento de Wagner-Meerwein catalizado por BF3:[33]
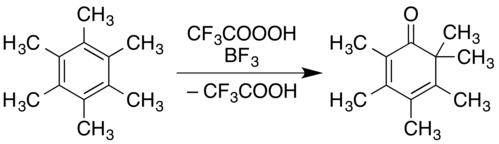
En cuanto a los compuestos aromáticos, un ejemplo demostrado en un informe de Organic Syntheses es la conversión de hexametilbenceno en 2,3,4,5,6,6-hexametil-2,4-ciclohexadienona:[9]
Corte oxidativo de arenos
Además de la simple oxidación de los anillos aromáticos para formar compuestos carbonílicos (véase § Oxidación con reordenamiento ácido), el ácido trifluoroperacético puede cortar completamente los enlaces carbono-carbono dentro del anillo. A diferencia de otras oxidaciones de estructuras alquilaromáticas, que producen ácidos benzoicos y compuestos relacionados mediante la escisión de la cadena alquílica en la posición bencilica reactiva, el ácido trifluoroperacético provoca una «oxidación inversa», escindiendo el propio anillo aromático y dejando intacto el grupo alquilo.[34][35]
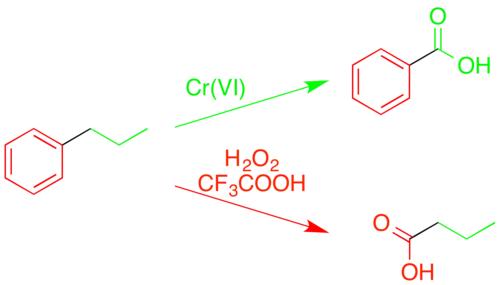
Esta selectividad para ciertos tipos de enlaces permite utilizarlo para descomponer mezclas complejas de hidrocarburos, como el carbón, con el fin de determinar detalles estructurales.[36][34]
Los sistemas aromáticos que contienen heteroátomos son resistentes a esta apertura del anillo, ya que la oxidación de los heteroátomos se produce de forma preferente y desactiva el anillo frente al ataque electrófilo del ácido peroxi. Por ejemplo, las purinas, las piridinas y las quinolinas forman N-óxidos,[5]mientras que los sistemas de azufre como el octafluorodibenzotiofeno se convierten en sulfones.[7][37]
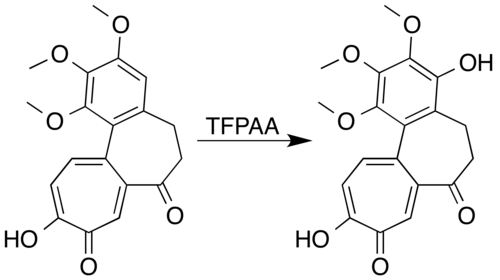
Los sistemas aromáticos con sustituyentes activadores del anillo pueden oxidarse para formar fenoles en lugar de sufrir una reacción de apertura del anillo. El mesitileno, por ejemplo, reacciona con el ácido trifluoroperacético para formar mesitol (2,4,6-trimetilfenol).[7] Los investigadores que intentaron formar una lactona mediante la oxidación de Baeyer-Villiger de la 7-oxodeacetamidocolchicina no pudieron preparar el producto deseado, pero lograron la oxidación del anillo aromático para producir un derivado fenólico con un alto rendimiento:[38]
Referencias
- ↑ a b Kürti, László; Czakó, Barbara (2005). Strategic Applications of Named Reactions in Organic Synthesis. Elsevier Academic Press. p. 28. ISBN 978-0-12-429785-2.
- ↑ a b c d e f Hiyama, Tamejiro (2000). "8.2 Trifluoroacetic acid and Trifluoroperacetic acid". Organofluorine Compounds: Chemistry and Applications. Springer Science & Business Media. pp. 255–257. ISBN 978-3-662-04164-2.
- ↑ a b Kataoka, T.; Yoshimatsu, M. (1995). "Alkyl Chalcogenides: Selenium- and Tellurium-based Functional Groups". In Ley, Steven V. (ed.). Synthesis: Carbon with One Heteroatom Attached by a Single Bond. Comprehensive Organic Functional Group Transformations. Elsevier. pp. 277–296. ISBN 978-0-08-042323-4.
- ↑ a b Carey, Francis A.; Sundberg, Richard J. (2007). "5.5 Addition Reactions Involving Epoxides". Advanced Organic Chemistry: Part A: Structure and Mechanisms (5th ed.). Springer Science & Business Media. pp. 503–514. ISBN 978-0-387-44897-8.
- ↑ a b c d e f g h i j k l m n ñ o p q Caster, Kenneth C.; Rao, A. Somasekar; Mohan, H. Rama; McGrath, Nicholas A.; Brichacek, Matthew (2012). "Trifluoroperacetic Acid". Encyclopedia of Reagents for Organic Synthesis. e-EROS Encyclopedia of Reagents for Organic Synthesis. doi:10.1002/047084289X.rt254.pub2. ISBN 978-0-471-93623-7.
- ↑ a b Freeman, Jeremiah P. (14/11/ 2002). "William D. Emmons: November 18, 1924 – December 8, 2001" (PDF). Org. Synth. 80: xxvii–xxix. Archivado del original (PDF) Marzo 16, 2015. Consultado 21/10/ 2017.
- ↑ a b c d e f g h Chambers, Richard D. (2004). "Functional Compounds Containing Oxygen, Sulphur or Nitrogen and their Derivatives". Fluorine in Organic Chemistry. CRC Press. pp. 242–243. ISBN 978-0-8493-1790-3.
- ↑ a b Luxon, S. G. (1992). Hazards in the Chemical Laboratory (5th ed.). Royal Society of Chemistry. p. 627. ISBN 978-0-85186-229-3.
- ↑ a b c Hart, Harold; Lange, Richard M.; Collins, Peter M. (1968). "2,3,4,5,6,6-Hexamethyl-2,4-cyclohexadien-1-one". Organic Syntheses. 48: 87. doi:10.15227/orgsyn.048.0087; Collected Volumes, vol. 5, p. 598.
- ↑ Cooper, Mark S.; Heaney, Harry; Newbold, Amanda J.; Sanderson, William R. (1990). "Oxidation Reactions Using Urea–Hydrogen Peroxide; A Safe Alternative to Anhydrous Hydrogen Peroxide". Synlett. 1990 (9): 533–535. doi:10.1055/s-1990-21156. S2CID 196724921.
- ↑ a b c d Ziegler, Fredrick E.; Metcalf, Chester A.; Nangia, Ashwini; Schulte, Gayle (1993). "Structure and total synthesis of sporol and neosporol". J. Am. Chem. Soc. 115 (7): 2581–2589. Bibcode:1993JAChS.115.2581Z. doi:10.1021/ja00060a006.
- ↑ Kang, Ho-Jung; Jeong, Hee-Sun (1996). "New Method of Generating Trifluoroperoxyacetic acid for the Baeyer-Villiger Reaction". Bull. Korean Chem. Soc. 17 (1): 5–6.
- ↑ a b Emmons, William D.; Ferris, Arthur F. (1953). "Oxidation Reactions with Pertrifluoroacetic Acid". J. Am. Chem. Soc. 75 (18): 4623–4624. Bibcode:1953JAChS..75.4623E. doi:10.1021/ja01114a539.
- ↑ a b Emmons, William D. (1954). "Peroxytrifluoroacetic Acid. I. The Oxidation of Nitrosamines to Nitramines". J. Am. Chem. Soc. 76 (13): 3468–3470. Bibcode:1954JAChS..76.3468E. doi:10.1021/ja01642a029.
- ↑ a b Emmons, William D. (1954). "Peroxytrifluoroacetic Acid. II. The Oxidation of Anilines to Nitrobenzenes". J. Am. Chem. Soc. 76 (13): 3470–3472. Bibcode:1954JAChS..76.3470E. doi:10.1021/ja01642a030.
- ↑ Emmons, William D.; Pagano, Angelo S.; Freeman, Jeremiah P. (1954). «Peroxytrifluoroacetic Acid. III. The Hydroxylation of Olefins». J. Am. Chem. Soc. 76 (13): 3472-3474. Bibcode:1954JAChS..76.3472E. doi:10.1021/ja01642a031. Emmons, William D.; Pagano, Angelo S. (1955). «Peroxytrifluoroacetic Acid. IV. The Epoxidation of Olefins». J. Am. Chem. Soc. 77 (1): 89-92. Bibcode:1955JAChS..77...89E. doi:10.1021/ja01606a029. Emmons, William D.; Lucas, George B. (1955). «Peroxytrifluoroacetic Acid. V. The Oxidation of Ketones to Esters». J. Am. Chem. Soc. 77 (8): 2287-2288. Bibcode:1955JAChS..77.2287E. doi:10.1021/ja01613a077. Emmons, William D.; Pagano, Angelo S. (1955). «Peroxytrifluoroacetic Acid. VI. The Oxidation of Oximes to Nitroparaffins». J. Am. Chem. Soc. 77 (17): 4557-4559. Bibcode:1955JAChS..77.4557E. doi:10.1021/ja01622a036.
- ↑ a b Aubé, Jeffrey; Fehl, Charlie; Liu, Ruzhang; McLeod, Michael C.; Motiwala, Hashim F. (1993). "6.15 Hofmann, Curtius, Schmidt, Lossen, and Related Reactions". Heteroatom Manipulations. Comprehensive Organic Synthesis II. Vol. 6. pp. 598–635. doi:10.1016/B978-0-08-097742-3.00623-6. ISBN 978-0-08-097743-0.
- ↑ Almond, M. R.; Stimmel, J. B.; Thompson, E. A.; Loudon, G. M. (1988). "Hofmann Rearrangement Under Mildly Acidic Conditions Using [I,I-Bis(Trifluoroacetoxy)]Iodobenzene: Cyclobutylamine Hydrochloride from Cyclobutanecarboxamide". Organic Syntheses. 66: 132. doi:10.15227/orgsyn.066.0132; Collected Volumes, vol. 8, p. 132.
- ↑ Myers, Andrew G. "Chemistry 115 Handouts: Oxidation" (PDF). Harvard University. Archivado del original (PDF) 16 Mayo 2017. Consultado 10/01/2017.
- ↑ Carruthers, William (1971). "6.3 Oxidation of Olefins". Some Modern Methods of Organic Synthesis. Cambridge University Press. pp. 259–280. ISBN 978-0-521-09643-0.
- ↑ Carruthers, William (1971). "6.5 Baeyer–Villiger oxidation of ketones". Some Modern Methods of Organic Synthesis. Cambridge University Press. pp. 287–290. ISBN 978-0-521-09643-0.
- ↑ a b Hagen, Timothy J. (2007). "Prilezhaev reaction". In Li, Jie Jack; Corey, E. J. (eds.). Name Reactions of Functional Group Transformations. John Wiley & Sons. pp. 274–281. ISBN 978-0-470-17650-4.
- ↑ Prileschajew, Nikolaus (1909). "Oxydation ungesättigter Verbindungen mittels organischer Superoxyde" [Oxidation of unsaturated compounds by means of organic superoxides]. Ber. Dtsch. Chem. Ges. (in German). 42 (4): 4811–4815. doi:10.1002/cber.190904204100.
- ↑ Hayakawa, Y.; Baba, Y.; Makino, S.; Noyori, R. (1978). "Carbon-carbon bond formation promoted by transition metal carbonyls. 19. General synthesis of tropane alkaloids via the polybromo ketone-iron carbonyl reaction". J. Am. Chem. Soc. 100 (6): 1786–1791. Bibcode:1978JAChS.100.1786H. doi:10.1021/ja00474a021.
- ↑ a b Pirrung, Michael C.; Morehead, Andrew T.; Young, Bruce G., eds. (2000). "10. Neosporol, Sporol". Part B: Bicyclic and Tricyclic Sesquiterpenes. The Total Synthesis of Natural Products. Vol. 11. John Wiley & Sons. pp. 222–224. ISBN 978-0-470-12963-0.
- ↑ Brooke, G. M.; Burdon, J.; Tatlow, J. C. (1961). "Aromatic polyfluoro-compounds. Part VII. The reaction of pentafluoronitrobenzene with ammonia". J. Chem. Soc.: 802–807. doi:10.1039/JR9610000802.
- ↑ Williams, W. Michael; Dolbier, William R. (1969). "Thermal and photochemical rearrangements of azine oxides. I. Pyrolytic decomposition to nitriles". J. Org. Chem. 34 (1): 155–157. doi:10.1021/jo00838a034.
- ↑ Taylor, Edward C.; McKillop, Alexander (1965). "A New Synthesis of 5-Nitropyrimidines". J. Org. Chem. 30 (9): 3153–3155. doi:10.1021/jo01020a067.
- ↑ Taylor, P. C. (1995). "Vinyl and Aryl Chalcogenides: Sulfur-, Selenium- and Tellurium-based Functional Groups". In Ley, Steven V. (ed.). Synthesis: Carbon with One Heteroatom Attached by a Single Bond. Comprehensive Organic Functional Group Transformations. Elsevier. pp. 705–736. ISBN 978-0-08-042323-4.
- ↑ Page, P. C. B.; Wilkes, R. D.; Reynolds, D. (1995). "Alkyl Chalcogenides: Sulfur-based Functional Groups". In Ley, Steven V. (ed.). Synthesis: Carbon with One Heteroatom Attached by a Single Bond. Comprehensive Organic Functional Group Transformations. Elsevier. pp. 113–276. ISBN 978-0-08-042323-4.
- ↑ a b c d e Treiber, Alexander (2002). "Mechanism of the Aromatic Hydroxylation of Thiophene by Acid-Catalyzed Peracid Oxidation". J. Org. Chem. 67 (21): 7261–7266. doi:10.1021/jo0202177. PMID 12375952.
- ↑ Anslyn, Eric V.; Dougherty, Dennis A. (2006). "8.8 Miscellaneous Experiments for Studying Mechanism". Modern Physical Organic Chemistry. University Science Books. pp. 471–482. ISBN 978-1-891389-31-3.
- ↑ Hart, Harold; Lerner, Lawrence R. (1967). "Oxidations with peroxytrifluoroacetic acid-boron trifluoride. IX. Direct oxidation of alkenes to ketones using peroxytrifluoroacetic acid–boron fluoride". J. Org. Chem. 32 (9): 2669–2673. doi:10.1021/jo01284a004
- ↑ a b Deno, Norman C.; Greigger, Barbara A.; Stroud, Stephen G. (1978). "New method for elucidating the structures of coal". Fuel. 57 (8): 455–459. Bibcode:1978Fuel...57..455D. doi:10.1016/0016-2361(78)90153-9.
- ↑ Deno, Norman C.; Greigger, Barbara A.; Messer, Lauren A.; Meyer, Michael D.; Stroud, Stephen G. (1977). "Aromatic ring oxidation of alkylbenzenes". Tetrahedron Lett. 18 (20): 1703–1704. doi:10.1016/S0040-4039(01)93253-8.
- ↑ Deno, Norman C.; Curry, Kenneth W.; Greigger, Barbara A.; Jones, A. Daniel; Rakitsky, Walter G.; Smith, Karen A.; Wagner, Karen; Minard, Robert D. (1980). "Dihydroaromatic structure of Illinois No. 6 Monterey coal". Fuel. 59 (10): 694–698. Bibcode:1980Fuel...59..694D. doi:10.1016/0016-2361(80)90021-6.
- ↑ Chambers, R. D.; Cunningham, J. A.; Spring, D. J. (1968). "Polyfluoroaryl organometallic compounds. Part VIII. Synthesis of and nucleophilic substitution in octafluorodibenzofuran". J. Chem. Soc. C: 1560–1565. doi:10.1039/J39680001560.
- ↑ Berg, Ulf; Bladha, Håkan; Mpamposa, Konstantinos (2004). "Stereochemical variations on the colchicine motif. Peracid oxidation of thiocolchicone. Synthesis, conformation and inhibition of microtubule assembly". Org. Biomol. Chem. 2 (14): 2125–2130. doi:10.1039/B402840F. PMID 15254641.