Síndrome de Luján-Fryns
| Síndrome de Lujan-Fryns | ||
|---|---|---|
 Síndrome de Lujan-Fryns en un varón adulto joven, con rasgos que incluyen cara larga y estrecha y mentón hundido. | ||
| Especialidad | Genética médica | |
| Sinónimos | ||
| Retraso mental ligado al cromosoma X con hábito marfanoide, síndrome de Lujan [1][2][3] | ||
El síndrome de Luján-Fryns es un trastorno genético ligado al cromosoma X que causa discapacidad intelectual de leve a moderada y rasgos descritos como hábito marfanoide, en referencia a un grupo de características físicas similares a las que se encuentran en el síndrome de Marfan.[4][5] Estos rasgos incluyen una estatura alta y delgada y extremidades largas y esbeltas.[5]
El síndrome de Luján-Fryns también se asocia a psicopatología y anomalías del comportamiento, y presenta una serie de malformaciones que afectan al cerebro y al corazón.[6][7][8] El trastorno se hereda de forma dominante ligada al cromosoma X y se atribuye a una mutación de sentido erróneo en el gen MED12.[9] En la actualidad no existe ningún tratamiento o terapia para la disfunción del MED12 subyacente, y la causa exacta del trastorno sigue sin estar clara.[10]
Signos y síntomas
La discapacidad intelectual suele oscilar entre leve y moderada, pero también se han descrito casos graves.[11][12] Una anomalía cerebral relativamente común que se observa es la agenesia del cuerpo calloso, un error del desarrollo embrionario en el que el cuerpo calloso (una estructura del cerebro de los mamíferos compuesta por nervios que permite la comunicación entre los hemisferios cerebrales izquierdo y derecho) no está presente.[13][14] Entre una serie de efectos neurológicos adversos que a veces se asocian con la ausencia del cuerpo calloso, se ha demostrado que la discapacidad intelectual se produce en un porcentaje aproximado del 73%.[14] Sin embargo, no se ha sugerido una correlación entre la agenesia del cuerpo calloso y la discapacidad intelectual en este síndrome.[15]
Psiquiatría
La psicopatología y las anomalías conductuales relacionadas suelen observarse en el síndrome de Luján-Fryns y pueden tenerse en cuenta en el diagnóstico del trastorno.[13]El más común de ellos es un trastorno del espectro similar al autismo y este síndrome se considera uno de los diversos trastornos genéticos asociados al autismo.[13][16] Entre las alteraciones adicionales de la psicopatología con manifestaciones conductuales que se han observado se incluyen: comportamiento psicótico,[17] esquizofrenia,[18] hiperactividad y trastorno por déficit de atención con hiperactividad,[15][19] agresividad,[19] trastorno negativista desafiante,[15][20] trastorno obsesivo-compulsivo,[15] timidez extrema,[19] problemas de aprendizaje,[15] deterioro cognitivo,[15] déficit de memoria a corto plazo,[15] baja tolerancia a la frustración,[15] disfunción social,[15] falta de control de los impulsos,[15] trastorno alimentario y malnutrición asociada, atribuida a la pérdida psicógena del apetito;[6] y piromanía. [13][15][20]
Aunque es de esperar que se produzcan afecciones psiquiátricas como éstas en el síndrome de Luján-Fryns, también se han dado casos de este trastorno con cierta preservación de las capacidades mentales y conductuales, como la resolución de problemas, el razonamiento y la inteligencia normal.[21]
La psicopatología de este trastorno suele mostrar esquizofrenia.[18] Cuando se diagnostica esquizofrenia a una persona con discapacidad intelectual, se debe considerar el síndrome de Luján-Fryns en el diagnóstico diferencial, tras confirmar la causa mediante métodos de evaluación psiquiátrica y genética adecuados.[18]
Hábito marfanoide
El síndrome se distingue clínicamente de otras formas de discapacidad intelectual ligadas al cromosoma X por la presencia de un hábito marfanoide.[11] El hábito marfanoide se caracteriza por un grupo de rasgos físicos comunes al síndrome de Marfan.[5] Además del síndrome de Marfan y el síndrome de Luján-Fryns, también se han observado rasgos marfanoides de este tipo en otros trastornos, uno de los cuales es la neoplasia endocrina múltiple de tipo 2.[22]
En este caso, los rasgos específicos identificados como marfanoides incluyen: cara larga y estrecha;[5][10] estatura alta y delgada;[9][10] extremidades largas y delgadas, dedos de manos y pies (no muy diferente a la aracnodactilia)[9][23][24] con hiperextensibilidad articular,[19] halluces acortados (los dedos gordos) y segundos dedos largos.[10]
El diagnóstico del hábito marfanoide suele retrasarse porque muchos de los rasgos físicos y características asociados a él no suelen ser evidentes hasta la adolescencia.[25]
Cabeza y cara
Las características craneofaciales y de otro tipo del síndrome de Luján-Fryns incluyen: hipoplasia maxilar (subdesarrollo del hueso maxilar superior),[10] una mandíbula pequeña (hueso maxilar inferior) y mentón retraído,[26][19] un paladar (el techo de la boca) de arco alto, con apiñamiento y desalineación de los dientes superiores;[5][7] macrocefalia (cráneo agrandado) con una frente prominente,[9][10] habla (voz) hipernasal,[5][7] una nariz larga con un puente nasal alto y estrecho;[10] un surco nasolabial profundo y corto (la hendidura en el labio superior, debajo de la nariz),[10] orejas de implantación baja con cierta retroversión aparente,[10] hipotonía (tono muscular disminuido),[9] pectus excavatum (una malformación del tórax),[10] tamaño testicular ligeramente aumentado hasta lo normal en los varones,[10][19] y convulsiones.[10]
El habla hipernasal, o "hipernasalidad", es principalmente el resultado de la insuficiencia velofaríngea, una aberración a veces congénita en la que el esfínter velofaríngeo deja pasar demasiado aire a la cavidad nasal durante el habla.[27][28] En el síndrome de Luján-Fryns, la hipernasalidad también puede deberse a que el paladar blando y la úvula no alcanzan la pared posterior de la faringe (la cavidad interior de la garganta donde generalmente se produce la deglución) durante el habla, una afección que puede estar asociada a un paladar hendido submucoso.[15][29]
Corazón
En varios casos del trastorno se han observado una serie de características que afectan al corazón, la más significativa de las cuales es la dilatación de la raíz aórtica, una sección de la aorta ascendente.[8] La dilatación (agrandamiento) de la raíz aórtica se asocia a un riesgo mucho mayor de disección de la pared aórtica, que da lugar a un aneurisma aórtico.[30] Dado que esta es una posible consecuencia potencialmente mortal, cuando se diagnostica por primera vez el trastorno se aplican métodos rutinarios de evaluación cardiaca, como el ecocardiograma, junto con resonancias magnéticas del cerebro para detectar la sospecha de agenesia del cuerpo calloso.[7] Otros efectos sobre el corazón que se han descrito son la comunicación interventricular y auricular.[8][19]
Causa
Se ha establecido que una mutaciones sin sentido en el gen MED12, situado en el cromosoma X humano, es la causa del síndrome de Luján-Fryns.[9][31] Las mutaciones sin sentido son mutaciones genéticas puntuales en las que un único nucleótido de la secuencia genética se intercambia con otro. Esto conduce a una sustitución errónea de un aminoácido concreto en la secuencia proteica durante la traducción. La mutación de sentido erróneo en el gen MED12, que causa el síndrome, se identifica como p.N1007S.[9] Esto indica que el aminoácido asparagina, normalmente situado en la posición 1007 de la secuencia MED12, ha sido sustituido por error por serina.[31] Esta mutación en MED12 provoca una expresión y actividad incorrectas de la proteína que codifica, lo que da lugar al trastorno.[9][10]
Fisiopatología
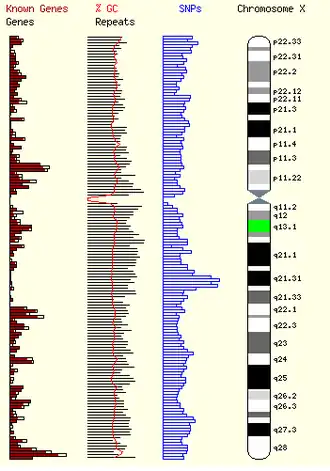
MED12, también conocida como «mediador de la transcripción de la ARN polimerasa II, subunidad 12 homóloga de S. cerevisiae», es una de varias subunidades del complejo mediador de mamíferos, que regula la ARN polimerasa II durante la transcripción del ARNm.[32][33]
El complejo mediador es necesario para la transcripción de la polimerasa II y actúa como puente entre la enzima polimerasa II y diferentes factores de transcripción específicos de un gen. El mediador puede contener hasta 30 subunidades, pero algunas de ellas solo son necesarias para regular la transcripción en determinados tejidos o células.[34] En la actualidad, no está claro el mecanismo exacto por el que la disfunción de MED12 da lugar al síndrome de Luján-Fryns y sus características neuropsicopáticas y físicas asociadas. El hábito marfanoide, el paladar muy arqueado y otras características de este trastorno pueden encontrarse en el síndrome de Marfan, un trastorno del tejido conectivo.[4] El hallazgo de dilatación de la raíz aórtica en ambos trastornos sugiere que una mutación en un gen regulador del tejido conectivo no especificado puede contribuir a la etiología del síndrome de Lujan-Fryns.[35][5][8][15]
Se han obtenido varios resultados experimentales interesantes estudiando las mutaciones de MED12 en el pez cebra, un modelo animal que representa a los vertebrados.[36][37][38] En este se descubrió que una mutación en MED12 era responsable del mutante inmóvil (mot). El pez cebra con la mutación mot presenta defectos neuronales y cardiovasculares, aunque no todos los tipos de neuronas están afectados. La introducción de ARNm de MED12 humano en el pez cebra restaura el desarrollo normal.[39] MED12 es también un coactivador crítico para el gen SOX9, implicado en la regulación del desarrollo de neuronas, cartílago y hueso. En el pez cebra, los defectos de MED12 provocan un mal desarrollo de estructuras embrionarias de vertebrados como la cresta neural, lo que alteraría la función de los sistemas nerviosos periférico y autónomo; y también provocan malformaciones de tipos celulares precursores de cartílago y hueso, como los osteocitos.[39][40][41] Algunas características encontradas en el síndrome de Luján-Fryns, como la agenesia del cuerpo calloso y las anomalías craneofaciales relacionadas con el cartílago, son similares a los defectos encontrados en el pez cebra con MED12 y mutaciones asociadas.[9]
Genética
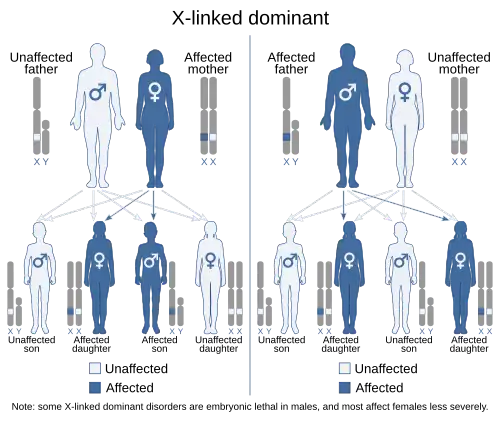
El síndrome de Luján-Fryns se hereda de forma dominante ligada al cromosoma X.[10][15][42] Esto significa que el gen defectuoso responsable del trastorno (MED12) está localizado en el cromosoma X, por lo que solo es necesario heredar una copia del gen defectuoso para sufrir el trastorno cuando se recibe de un progenitor portador. Normalmente, los varones son hemicigotos para el cromosoma X, es decir, solo tienen una copia. Como resultado, los trastornos dominantes ligados al cromosoma X suelen mostrar una mayor expresividad en los varones que en las mujeres. Se cree que este fenómeno ocurre con el síndrome de Luján-Fryns.[15][42]
Dado que el cromosoma X es uno de los cromosomas sexuales (el otro es el cromosoma Y), la herencia ligada al cromosoma X viene determinada por el cariotipo del progenitor portador de un gen específico y a menudo puede parecer compleja. Esto se debe a que, normalmente, las mujeres tienen dos copias del cromosoma X, mientras que los hombres solo tienen una copia. La diferencia entre los patrones de herencia dominante y recesiva también influye a la hora de determinar las probabilidades de que un niño herede un trastorno ligado al cromosoma X de sus padres.
En el síndrome de Luján-Fryns, se sospechó de una herencia dominante ligada al cromosoma X, ya que los hermanos de una familia presentaban ambos el trastorno.[15][42] Un escenario como éste también sería posible con una herencia recesiva ligada al cromosoma X, pero en este caso concreto, se creía que la niña era un heterocigoto manifiesto[15][42] portador de una copia del gen mutado.
También se han descrito casos esporádicos, en los que el trastorno está presente en un individuo sin antecedentes familiares, en un pequeño número de varones afectados.[15][17][43]
Similitudes con otras enfermedades genéticas
Se descubrió que un individuo que presentaba discapacidad intelectual y otros síntomas similares al síndrome tenía una deleción terminal de la región subtelomérica en el brazo corto del cromosoma 5.[29] La deleción de esta zona del cromosoma 5 se asocia a discapacidad intelectual, comportamiento psicótico, autismo, macrocefalia y habla hipernasal, así como al trastorno del síndrome de Cri du chat.[29][44] Fryns (2006) sugiere que se realice un examen detallado del cromosoma 5 con FISH como parte del diagnóstico diferencial del síndrome de Luján-Fryns.[10]
Las mutaciones en el gen UPF3B, que también se encuentra en el cromosoma X, son otra causa de discapacidad intelectual ligada al cromosoma X.[45] UPF3B forma parte del complejo de descomposición del ARNm mediada por mutaciones sin sentido (NMD), que realiza la vigilancia del ARNm, detectando secuencias de ARNm que han sido erróneamente truncadas (acortadas) por la presencia de mutaciones sin sentido.[46] Las mutaciones en UPF3B alteran e impiden el funcionamiento normal de la vía NMD, lo que da lugar a la traducción y expresión de secuencias de ARNm truncadas en proteínas disfuncionales que pueden estar asociadas a errores de desarrollo y discapacidad intelectual.[46][47] Se descubrió que individuos de dos familias diagnosticadas de síndrome de Luján-Fryns y de una familia con síndrome FG tenían mutaciones en UPF3B, lo que confirma que las presentaciones clínicas de las diferentes mutaciones pueden solaparse.[47]
Diagnóstico
Aunque suele sospecharse la presencia del síndrome de Luján-Fryns cuando se observan conjuntamente discapacidad intelectual y hábito marfanoide en un paciente, el diagnóstico de este puede confirmarse por la presencia de la mutación de sentido erróneo p.N1007S en el gen MED12.[9][10][11]
Diagnóstico diferencial
En el diagnóstico diferencial del síndrome de Luján-Fryns, otro trastorno que presenta algunas características y síntomas que también se asocia a una mutación de sentido erróneo de MED12 es el síndrome de Opitz-Kavea (FGS).[9][48] Las características comunes que comparten tanto el síndrome de Luján-Fryns como el FGS incluyen discapacidad intelectual ligada al cromosoma X, hiperactividad, macrocefalia, agenesia del cuerpo calloso e hipotonía.[9] Algunas características notables del FGS que no se han descrito en el síndrome de Lujan-Fryns son la locuacidad excesiva, la fuerza constante en las habilidades de socialización, el ano imperforado (oclusión del ano) y el hipertelorismo ocular (ojos extremadamente separados).[49][50]
Mientras que el síndrome de Luján-Fryns se asocia a la mutación de sentido erróneo p.N1007S, el FGS se asocia a la mutación de sentido erróneo p.R961W.[9][51] Dado que ambos trastornos se originan a partir de un tipo idéntico de mutación en el mismo gen, aunque presentan características similares pero distintas, se considera que el síndrome de Lujan-Fryns y el FGS son alélicos.[9][10][15][48] En el contexto de MED12, esto sugiere que el fenotipo de cada trastorno está relacionado con la forma en que sus respectivas mutaciones alteran la secuencia MED12 y su función.[9][31][48]
Tratamiento
Aunque no existe un tratamiento específico para la causa genética subyacente del trastorno, pueden considerarse procedimientos correctivos, medidas de intervención preventiva y terapias en el tratamiento y la gestión de los numerosos problemas craneofaciales, ortopédicos y psiquiátricos asociados al trastorno. Los problemas más graves, como la afectación cardiaca o los ataques epilépticos, deben examinarse y controlarse de forma rutinaria. Para diagnosticar y prevenir los trastornos psiquiátricos y los problemas de conducta relacionados, como la psicosis y los arrebatos de agresividad, debe prestarse una atención especial y un seguimiento especializado, que incluya métodos y terapias de evaluación neuropsicológica y una educación especial.[10]
Epidemiología
El síndrome de Luján-Fryns es un síndrome raro dominante ligado al cromosoma X y es más frecuente en varones que en mujeres. Aún no se ha determinado su prevalencia en la población general.[10]
Historia
El síndrome de Luján-Fryns debe su nombre a los médicos J. Enrique Luján y Jean-Pierre Fryns.[23] La observación inicial de una presunta discapacidad intelectual ligada al cromosoma X con rasgos marfanoides y efectos craneofaciales como un paladar de arco alto fue descrita por Luján en 1984.[19] En el informe se señalaban cuatro varones afectados de una familia numerosa (consanguínea)[9][15][19] A partir de 1987, Fryns informaron de investigaciones adicionales de discapacidad intelectual combinada ligada al cromosoma X y hábito marfanoide en otras familias, incluidos dos hermanos[5] El trastorno pronto se conoció como síndrome de Luján-Fryns.[42]
Véase también
Referencias
- ↑ Lacombe, D.; Bonneau, D.; Verloes, A.; Couet, D.; Koulischer, L.; Battin, J. (1993). «Lujan-Fryns syndrome (X-linked mental retardation with marfanoid habitus): report of three cases and review». Genetic Counseling (Geneva, Switzerland) (en inglés) 4 (3): 193-198. ISSN 1015-8146. PMID 8267926.
- ↑ Fryns, J. P.; Van Den Berghe, H. (1991). «X-linked mental retardation with Marfanoid habitus: a changing phenotype with age?». Genetic Counseling (Geneva, Switzerland) (en inglés) 2 (4): 241-244. ISSN 1015-8146. PMID 1799424.
- ↑ Schwartz, C. E.; Tarpey, P. S.; Lubs, H. A.; Verloes, A.; May, M. M.; Risheg, H.; Friez, M. J.; Futreal, P. A. (Julio 2007). «The original Lujan syndrome family has a novel missense mutation (p.N1007S) in the MED12 gene». Journal of Medical Genetics (en inglés) 44 (7): 472-477. ISSN 0022-2593. PMC 2597996. PMID 17369503. doi:10.1136/jmg.2006.048637.
- ↑ a b «MARFAN SYNDROME; MFS». Online Mendelian Inheritance in Man (en inglés).
- ↑ a b c d e f g h Fryns, J. P.; Buttiens, M.; Opitz, J. M.; Reynolds, J. F. (Octubre 1987). «X-linked mental retardation with marfanoid habitus». American Journal of Medical Genetics (en inglés) 28 (2): 267-274. ISSN 0148-7299. PMID 3322000. doi:10.1002/ajmg.1320280202.
- ↑ a b Alonso, P.; Pintos, G.; Almazan, F.; Hernández, L.; Loran, E.; Menchon, J. M.; Vallejo, J. (Julio 2006). «Eating disorder in a patient with phenotypical features of Lujan-Fryns syndrome». Clinical Dysmorphology (en inglés) 15 (3): 181-184. ISSN 0962-8827. PMID 16760741. doi:10.1097/01.mcd.0000220610.24908.a4. 7415391.
- ↑ a b c d Lerma-Carrillo, I.; Molina, J. D.; Cuevas-Duran, T.; Julve-Correcher, C.; Espejo-Saavedra, J. M.; Andrade-Rosa, C.; Lopez-Muñoz, F. (Diciembre 2006). «Psychopathology in the Lujan-Fryns syndrome: report of two patients and review». American Journal of Medical Genetics Part A (en inglés) 140 (24): 2807-2811. ISSN 1552-4825. PMID 17036352. doi:10.1002/ajmg.a.31503. 22491132.
- ↑ a b c d Wittine, L. M.; Josephson, K. D.; Williams, M. S. (Octubre 1999). «Aortic root dilation in apparent Lujan-Fryns syndrome». American Journal of Medical Genetics (en inglés) 86 (5): 405-409. ISSN 0148-7299. PMID 10508979. doi:10.1002/(SICI)1096-8628(19991029)86:5<405::AID-AJMG2>3.0.CO;2-1.
- ↑ a b c d e f g h i j k l m n ñ o Schwartz, C. E; Tarpey, P. S; Lubs, H. A; Verloes, A; May, M. M; Risheg, H.; Friez, M. J.; Futreal, P. A. et al. (Julio 2007). «The original Lujan syndrome family has a novel missense mutation (p.N1007S) in the MED12 gene». Journal of Medical Genetics (en inglés) 44 (7): 472-477. ISSN 0022-2593. PMC 2597996. PMID 17369503. doi:10.1136/jmg.2006.048637.
- ↑ a b c d e f g h i j k l m n ñ o p q r Buggenhout, G. V.; Fryns, J. -P. (Julio 2006). «Lujan-Fryns syndrome (mental retardation, X-linked, marfanoid habitus)». Orphanet Journal of Rare Diseases (en inglés) 1: 26. PMC 1538574. PMID 16831221. doi:10.1186/1750-1172-1-26.
- ↑ a b c Fryns, J. P.; Buttiens, M.; Van Den Berghe, H. (Enero 1988). «Chromosome X-linked mental retardation and marfanoid syndrome». Journal de Génétique Humaine (en inglés) 36 (1–2): 123-128. ISSN 0021-7743. PMID 3379374.
- ↑ Mégarbané A, C. C.; Chammas, C. (1997). «Severe mental retardation with marfanoid habitus in a young Lebanese male. A diagnostic challenge». Genetic Counseling (Geneva, Switzerland) (en inglés) 8 (3): 195-200. ISSN 1015-8146. PMID 9327261.
- ↑ a b c d Lerma-Carrillo, I.; Molina, J. D.; Cuevas-Duran, T.; Julve-Correcher, C.; Espejo-Saavedra, J. M.; Andrade-Rosa, C.; Lopez-Muñoz, F. (Diciembre 2006). «Psychopathology in the Lujan-Fryns syndrome: report of two patients and review». American Journal of Medical Genetics Part A (en inglés) 140 (24): 2807-2811. ISSN 1552-4825. PMID 17036352. doi:10.1002/ajmg.a.31503. 22491132.
- ↑ a b Jeret, J. S.; Serur, D.; Wisniewski, K. E.; Lubin, R. A. (1987). «Clinicopathological findings associated with agenesis of the corpus callosum». Brain & Development (en inglés) 9 (3): 255-264. ISSN 0387-7604. PMID 3310713. doi:10.1016/s0387-7604(87)80042-6. 4761497.
- ↑ a b c d e f g h i j k l m n ñ o p q r s «INTELLECTUAL DEVELOPMENTAL DISORDER, X-LINKED, SYNDROMIC, LUJAN-FRYNS TYPE; MRXSLF». Online Mendelian Inheritance in Man (en inglés).
- ↑ Artigas-Pallarés, J.; Gabau-Vila, E.; Guitart-Feliubadaló, M. (Enero 2005). «Syndromic autism: II. Genetic syndromes associated with autism». Revista de Neurología (en inglés) 40 (Suppl 1): S151-S162. ISSN 0210-0010. PMID 15736079. doi:10.33588/rn.40S01.2005073.
- ↑ a b Lalatta, F.; Livini, E.; Selicorni, A.; Briscioli, V.; Vita, A.; Lugo, F.; Zollino, M.; Gurrieri, F. et al. (Febrero 1991). «X-linked mental retardation with marfanoid habitus: first report of four Italian patients». American Journal of Medical Genetics (en inglés) 38 (2–3): 228-232. ISSN 0148-7299. PMID 2018063. doi:10.1002/ajmg.1320380211.
- ↑ a b c De Hert, M.; Steemans, D.; Theys, P.; Fryns, J. P.; Peuskens, J. (Abril 1996). «Lujan-Fryns syndrome in the differential diagnosis of schizophrenia». American Journal of Medical Genetics (en inglés) 67 (2): 212-213. PMID 8723050. doi:10.1002/(SICI)1096-8628(19960409)67:2<212::AID-AJMG13>3.0.CO;2-M.
- ↑ a b c d e f g h i Lujan, J. E.; Carlin, M. E.; Lubs, H. A.; Opitz, J. M. (Enero 1984). «A form of X-linked mental retardation with marfanoid habitus». American Journal of Medical Genetics (en inglés) 17 (1): 311-322. ISSN 0148-7299. PMID 6711603. doi:10.1002/ajmg.1320170124.
- ↑ a b Williams, M. S. (Diciembre 2006). «Neuropsychological evaluation in Lujan-Fryns syndrome: commentary and clinical report». American Journal of Medical Genetics Part A (en inglés) 140 (24): 2812-2815. ISSN 1552-4825. PMID 17103446. doi:10.1002/ajmg.a.31501.
- ↑ Donders, J.; Toriello, H.; Van Doornik, S. (Enero 2002). «Preserved neurobehavioral abilities in Lujan-Fryns syndrome». American Journal of Medical Genetics (en inglés) 107 (3): 243-246. ISSN 0148-7299. PMID 11807907. doi:10.1002/ajmg.10144.
- ↑ Prabhu, M.; Khouzam, R. N.; Insel, J. (Noviembre 2004). «Multiple endocrine neoplasia type 2 syndrome presenting with bowel obstruction caused by intestinal neuroma: case report». Southern Medical Journal (en inglés) 97 (11): 1130-1132. ISSN 0038-4348. PMID 15586612. doi:10.1097/01.SMJ.0000140873.29381.12.
- ↑ a b «Lujan-Fryns syndrome». Who Named It? (en inglés).
- ↑ Buntinx, I. M.; Willems, P. J.; Spitaels, S. E.; Van Reempst, P. J.; De Paepe, A. M.; Dumon, J. E. (Abril 1991). «Neonatal Marfan syndrome with congenital arachnodactyly, flexion contractures, and severe cardiac valve insufficiency». Journal of Medical Genetics (en inglés) 28 (4): 267 - 273. ISSN 0022-2593. PMC 1016831. PMID 1856834. doi:10.1136/jmg.28.4.267.
- ↑ Fryns, J. P.; Van Den Berghe, H. (1991). «X-linked mental retardation with Marfanoid habitus: a changing phenotype with age?». Genetic Counseling (en inglés) (Ginebra (Suiza)) 2 (4): 241-244. ISSN 1015-8146. PMID 1799424.
- ↑ Schwartz, C. E; Tarpey, P. S.; Lubs, H. A.; Verloes, A.; May, M. M.; Risheg, H.; Friez, M. J.; Futreal, P. A. et al. (Julio de 2007). «The original Lujan syndrome family has a novel missense mutation (p.N1007S) in the MED12 gene». Journal of Medical Genetics (en inglés) 7 (44): 472-477. ISSN 0022-2593. PMC 2597996. PMID 17369503. doi:10.1136/jmg.2006.048637.
- ↑ Willging, J. P. (Octubre 1999). «Velopharyngeal insufficiency». International Journal of Pediatric Otorhinolaryngology (en inglés) 49 (1): 307-309. ISSN 0165-5876. PMID 10577827. doi:10.1016/S0165-5876(99)00182-2.
- ↑ Warren, D. W.; Dalston, R. M.; Mayo, R. (Julio 1994). «Hypernasality and velopharyngeal impairment». The Cleft Palate-Craniofacial Journal (en inglés) 31 (4): 257-262. ISSN 1055-6656. PMID 7918520. doi:10.1597/1545-1569(1994)031<0257:HAVI>2.3.CO;2.
- ↑ a b c Stathopulu, E.; Ogilvie, C. M.; Flinter, F. A. (Junio 2003). «Terminal deletion of chromosome 5p in a patient with phenotypical features of Lujan-Fryns syndrome». American Journal of Medical Genetics Part A (en inglés) 3 (119): 363-366. ISSN 1552-4825. PMID 12784307. doi:10.1002/ajmg.a.10268.
- ↑ Gambarin, F.; Favalli, V.; Serio, A.; Regazzi, M.; Pasotti, M.; Klersy, C.; Dore, R.; Mannarino, S. et al. (Abril 2009). «Rationale and design of a trial evaluating the effects of losartan vs. Nebivolol vs. The association of both on the progression of aortic root dilation in Marfan syndrome with FBN1 gene mutations». Journal of Cardiovascular Medicine (Hagerstown, Md.) (en inglés) 10 (4): 354-362. ISSN 1558-2027. PMID 19430350. doi:10.2459/JCM.0b013e3283232a45.
- ↑ a b c «Mediator Complex Subunit 12 ; Med12». Online Mendelian Inheritance in Man (en inglés).
- ↑ Biddick, R.; Young, E. (Septiembre 2005). «Yeast mediator and its role in transcriptional regulation». Comptes Rendus Biologies (en inglés) 328 (9): 773-782. ISSN 1631-0691. PMID 16168358. doi:10.1016/j.crvi.2005.03.004.
- ↑ Sims, R. J. 3rd; Mandal, S. S.; Reinberg, D. (Junio 2004). «Recent highlights of RNA-polymerase-II-mediated transcription». Current Opinion in Cell Biology (en inglés) 16 (3): 263-271. ISSN 0955-0674. PMID 15145350. doi:10.1016/j.ceb.2004.04.004.
- ↑ Malik, S.; Roeder, R. G. (Junio 2000). «Transcriptional regulation through Mediator-like coactivators in yeast and metazoan cells». Trends in Biochemical Sciences (en inglés) 25 (6): 277-283. ISSN 0968-0004. PMID 10838567. doi:10.1016/S0968-0004(00)01596-6.
- ↑ Lacombe, D.; Bonneau, D.; Verloes, A.; Couet, D.; Koulischer, L.; Battin, J. (1993). «Lujan-Fryns syndrome (X-linked mental retardation with marfanoid habitus): report of three cases and review». Genetic Counseling (en inglés) (Ginebra (Suiza)) 4 (3): 193-198. ISSN 1015-8146. PMID 8267926.
- ↑ Chakraborty C, H. C.; Hsu, C. H.; Wen, Z. H.; Lin, C. S.; Agoramoorthy, G. (Febrero 2009). «Zebrafish: a complete animal model for in vivo drug discovery and development». Current Drug Metabolism (en inglés) 10 (2): 116-124. ISSN 1389-2002. PMID 19275547. doi:10.2174/138920009787522197.
- ↑ Kari, G.; Rodeck, U.; Dicker, A. P. (Julio 2007). «Zebrafish: an emerging model system for human disease and drug discovery». Clinical Pharmacology and Therapeutics (en inglés) 82 (1): 70-80. ISSN 0009-9236. PMID 17495877. doi:10.1038/sj.clpt.6100223.
- ↑ McGonnell, I. M.; Fowkes, R. C. (Junio 2006). «Fishing for gene function--endocrine modelling in the zebrafish» (PDF). The Journal of Endocrinology (en inglés) 189 (3): 425-439. ISSN 0022-0795. PMID 16731775. doi:10.1677/joe.1.06683.
- ↑ a b Wang, X.; Yang, N.; Uno, E.; Roeder, R. G.; Guo, S. (Noviembre 2006). «A subunit of the mediator complex regulates vertebrate neuronal development» (HTML). Proceedings of the National Academy of Sciences of the United States of America (Free full text) (en inglés) 103 (46): 17284-17289. Bibcode:2006PNAS..10317284W. ISSN 0027-8424. PMC 1859923. PMID 17088561. doi:10.1073/pnas.0605414103.
- ↑ Rau, M. J.; Fischer, S.; Neumann, C. J. (Agosto 2006). «Zebrafish Trap230/Med12 is required as a coactivator for Sox9-dependent neural crest, cartilage and ear development». Developmental Biology (en inglés) 296 (1): 83-93. ISSN 0012-1606. PMID 16712834. doi:10.1016/j.ydbio.2006.04.437.
- ↑ Hong, S. -K.; Haldin, C. E.; Lawson, N. D.; Weinstein, B. M.; Dawid, I. B.; Hukriede, N. A. (Diciembre 2005). «The zebrafish kohtalo/trap230 gene is required for the development of the brain, neural crest, and pronephric kidney». Proceedings of the National Academy of Sciences of the United States of America (en inglés) 102 (51): 18473-18478. Bibcode:2005PNAS..10218473H. ISSN 0027-8424. PMC 1311743. PMID 16344459. doi:10.1073/pnas.0509457102.
- ↑ a b c d e Gurrieri, F.; Neri, G. (Febrero 1991). «A girl with the Lujan-Fryns syndrome». American Journal of Medical Genetics (en inglés) 38 (2–3): 290-291. ISSN 0148-7299. PMID 2018074. doi:10.1002/ajmg.1320380225.
- ↑ Fryns, J. P. (Febrero 1991). «X-linked mental retardation with marfanoid habitus». American Journal of Medical Genetics (en inglés) 38 (2–3): 233. ISSN 0148-7299. PMID 2018064. doi:10.1002/ajmg.1320380212.
- ↑ Fang, J. S.; Lee, K. F.; Huang, C. T.; Syu, C. L.; Yang, K. J.; Wang, L. H.; Liao, D. L.; Chen, C. H. (Junio 2008). «Cytogenetic and molecular characterization of a three-generation family with chromosome 5p terminal deletion». Clinical Genetics (en inglés) 73 (6): 585-590. ISSN 0009-9163. PMID 18400035. doi:10.1111/j.1399-0004.2008.00995.x.
- ↑ «UPF3B REGULATOR OF NONSENSE-MEDIATED mRNA DECAY; UPF3B». Mendelian Inheritance in Man (en inglés).
- ↑ a b Chang, Y. F.; Imam, J. S.; Wilkinson, M. F. (2007). «The nonsense-mediated decay RNA surveillance pathway». Annual Review of Biochemistry (en inglés) 76: 51-74. ISSN 0066-4154. PMID 17352659. doi:10.1146/annurev.biochem.76.050106.093909.
- ↑ a b Tarpey, P. S.; Raymond, F. L.; Nguyen, L. S.; Rodriguez, J.; Hackett, A.; Vandeleur, L.; Smith, R.; Shoubridge, C. et al. (Septiembre 2007). «Mutations in UPF3B, a member of the nonsense-mediated mRNA decay complex, cause syndromic and nonsyndromic mental retardation». Nature Genetics (en inglés) 39 (9): 1127-1133. ISSN 1061-4036. PMC 2872770. PMID 17704778. doi:10.1038/ng2100.
- ↑ a b c «OPITZ-KAVEGGIA SYNDROME; OKS». Mendelian Inheritance in Man (en inglés).
- ↑ Graham, J. M.; Superneau, D.; Rogers, R. C.; Corning, K.; Schwartz, C. E.; Dykens, E. M. (1999). «Clinical and behavioral characteristics in FG syndrome». American Journal of Medical Genetics (en inglés) 85 (5): 470-475. PMID 10405444. doi:10.1002/(SICI)1096-8628(19990827)85:5<470::AID-AJMG7>3.0.CO;2-S.
- ↑ Graham, J. M. Jr.; Visootsak, J.; Dykens, E.; Huddleston, L.; Clark, R. D.; Jones, K. L.; Moeschler, J. B..; Opitz, J. M.. et al. (Diciembre 2008). «Behavior of 10 patients with FG Syndrome (Opitz-Kaveggia Syndrome) and the p.R961W Mutation in the MED12 Gene». American Journal of Medical Genetics Part A (en inglés). 146A (23): 3011-3017. ISSN 1552-4825. PMC 3092600. PMID 18973276. doi:10.1002/ajmg.a.32553.
- ↑ Risheg, H.; Graham, J. M.; Clark, R. D.; Rogers, R. C.; Opitz, J. M.; Moeschler, J. B.; Peiffer, A. P.; May, M. et al. (Abril 2007). «A recurrent mutation in MED12 leading to R961W causes Opitz-Kaveggia syndrome». Nature Genetics (en inglés) 39 (4): 451-453. ISSN 1061-4036. PMID 17334363. doi:10.1038/ng1992.
Bibliografía
- Van Buggenhout, G. J. C. M.; Trommelen, J. C. M.; Brunner, H. G.; Hamel, B. C. J.; Fryns, J. P. (Enero 2001). «The clinical phenotype in institutionalised adult males with X-linked mental retardation (XLMR)». Annales de Génétique. 1 (en iglés) 44: 47-55. ISSN 0003-3995. PMID 11334618.
Enlaces externos
- Entrada de GeneReview/NIH/UW sobre los trastornos relacionados con MED12 (en inglés)
 Síndrome de Luján-Fryns en Scholia
Síndrome de Luján-Fryns en Scholia
 Datos: Q640836
Datos: Q640836 Multimedia: Lujan–Fryns syndrome / Q640836
Multimedia: Lujan–Fryns syndrome / Q640836