Síndrome nasodigitoacústico
| Síndrome nasodigitoacústico / Síndrome de Keipert | ||
|---|---|---|
| Especialidad |
Pediatría Neurología | |
| Inicio habitual | Infancia | |
| Causas |
Genéticas: recesiva ligada al cromosoma X | |
| Diagnóstico | Clínico | |
| Tratamiento | Sintomático | |
| Frecuencia | 1 cada 2.000 en Europa | |
El síndrome nasodigitoacústico, también llamado síndrome de Keipert, es un síndrome congénito poco frecuente descrito por primera vez por J.A. Keipert y sus colegas en 1973. El síndrome se caracteriza por una nariz deforme, pulgares y dedos gordos anchos, braquidactilia, pérdida auditiva neurosensorial, rasgos faciales como hipertelorismo (ojos inusualmente separados) y retraso en el desarrollo.
Se cree que se hereda de forma recesiva ligada al cromosoma X, lo que significa que la mutación genética causante del trastorno se localiza en el cromosoma X, y mientras que para que una mujer nazca con el trastorno deben heredarse dos copias del gen mutado, basta con una copia para que un varón nazca con el trastorno. Es probable que el síndrome nasodigitoacústico esté causado por un gen mutado localizado en el cromosoma X entre las posiciones Xq22.2-q28.
No se ha determinado la incidencia del síndrome, pero se considera que afecta a menos de 200.000 personas en Estados Unidos y a no más de 1 por cada 2.000 en Europa. Es similar al síndrome de Keutel, Muenke, Rubinstein y Teunissen-Cremers.[1][2][3][4][5]
Características
El síndrome nasodigitoacústico es congénito y se caracteriza por una serie de rasgos nasales, faciales y craneales. Entre ellos, un puente nasal (parte superior de la nariz, entre los ojos) ancho y alto, a veces deprimido, y una punta nasal aplanada,[2][6][7] lo que puede dar a la nariz un aspecto acortado y arqueado.[8]También se ha observado hipertelorismo (ojos inusualmente separados),[4]huesos frontales prominentes y cresta supraorbitaria (la cresta de la ceja), pliegues epicánticos bilaterales (un colgajo extra de piel sobre los párpados), una frente ancha y un perímetro cefálico general agrandado. También se ha descrito un abombamiento del labio superior con una forma exagerada de arco de cupido,[9]e hipoplasia maxilar (subdesarrollo de la mandíbula superior) con retracción.[2][7][10]
Con el síndrome se han observado varias anomalías que afectan a los dedos de las manos y de los pies. Se ha descrito un ensanchamiento de los pulgares y los dedos gordos de los pies (aluces) en dos hermanos. El ensanchamiento era evidente en todas las falanges distales de los dedos, aunque los meñiques no estaban afectados y parecían clinodactilicos (deformados o doblados hacia los otros dedos)[2] Otros informes describen este ensanchamiento de los pulgares y los dedos gordos de los pies, con braquidactilia (acortamiento) en las falanges distales de los otros dedos excepto los meñiques en los individuos afectados. En las radiografías de un niño de dos años con este trastorno, se demostró que la braquidactilia estaba causada por el acortamiento de las epífisis (extremos de las articulaciones) de las falanges distales.[7][11] La anchura y la braquidactilia de los dedos gordos, en particular, pueden darles un aspecto atrofiado, redondeado y en forma de muñón.[8]
Las anomalías auditivas o "acústicas" observadas en el síndrome incluyen pérdida de audición neurosensorial y ronquera. Se han descrito dos hermanos turcos afectados con una forma leve de esta pérdida de audición y voz ronca. El examen laringoscópico de ambos hermanos reveló una inflamación de las cuerdas vocales y una epiglotis malformada.[6] También se observó hipoacusia neurosensorial y ronquera en una niña de 10 años y su padre,[10] así como en otros casos.[3][7]
Otras características del síndrome son retraso del desarrollo, retraso del crecimiento, estenosis pulmonar (obstrucción del flujo sanguíneo del ventrículo derecho del corazón a la arteria pulmonar) con disnea asociada y agenesia renal (falta de desarrollo de los riñones durante el periodo fetal). También se han observado testículos no descendidos, hiperactividad y comportamiento agresivo.[2][3][4]
Genética

Se cree que el síndrome nasodigitoacústico está causado por una mutación en un gen del cromosoma X. Un estudio de 2007 concluyó, basándose en el análisis de marcadores de microsatélites (pequeñas secuencias de genes que se encuentran en común entre individuos que tienen la misma etnia, ascendencia o enfermedad genética) de la familia descrita por Keipert, que este gen estaba probablemente localizado en el brazo largo del cromosoma X entre las posiciones Xq22.2-q28. Sin embargo, esto no es definitivo y no se ha nombrado ningún gen específico.[3]
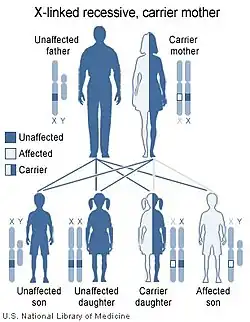
Se cree firmemente que el síndrome se hereda de forma recesiva ligada al cromosoma X.[3] Cuando una mujer es portadora de un gen mutado en una de sus dos copias del cromosoma X, tiene un 50% de probabilidades de transmitir la mutación a sus hijos. Al igual que ella, una hija que herede esta mutación será portadora, pero no padecerá la enfermedad asociada. Sin embargo, un hijo varón que herede la mutación tendrá la enfermedad; esto se debe a que los varones sólo tienen una copia del cromosoma X y, por tanto, sólo podrían expresar la mutación de la enfermedad.
Sin embargo, esta forma de herencia del síndrome nasodigitoacústico aún no es absoluta, ya que se ha descrito una niña con el trastorno. Se sugiere que son necesarios más análisis para establecer formalmente la herencia.[7][10]
Las mutaciones causantes de este síndrome se han identificado en el gen glypican 4 (GPC4),[12] situado en el brazo largo del cromosoma X (Xq26.2).
Diagnóstico
La constelación de anomalías observadas en el síndrome nasodigitoacústico da lugar a un diagnóstico diferenciado. Los criterios diagnósticos del trastorno son falanges distales anchas de los pulgares y los dedos gordos de los pies, acompañadas de una nariz ancha y acortada, pérdida auditiva neurosensorial y retraso del desarrollo, con predominio en varones.[4][9]
Clasificación
El síndrome nasodigitoacústico es similar a varios síndromes que comparten sus características.[4][7] La braquidactilia de las falanges distales, la sordera neurosensorial y la estenosis pulmonar son frecuentes en el síndrome de Keutel.[13]En el síndrome de Muenke, se han descrito retraso del desarrollo, braquidactilia distal y sordera neurosensorial; las características del síndrome de Teunissen-Cremers incluyen aberraciones nasales y anchura de los pulgares y los dedos gordos de los pies, también con braquidactilia. [14][15]Los pulgares y los dedos gordos de los pies anchos son las características principales del síndrome de Rubinstein.[16]
Tratamiento
Algunas de las características del síndrome nasodigitoacústico pueden controlarse o tratarse. La pérdida de audición neurosensorial en humanos puede deberse a la pérdida de células ciliadas (receptores sensoriales del oído interno asociados a la audición). Esto puede ser hereditario y/o dentro de un síndrome, como es el caso del síndrome nasodigitoacústico,[4] o atribuirse a infecciones como los virus. Para el tratamiento de la pérdida de audición neurosensorial se han utilizado audífonos. Los tratamientos, dependiendo de la causa y la gravedad, pueden incluir un enfoque farmacológico (es decir, el uso de ciertos esteroides) o una intervención quirúrgica, como un implante coclear.[17][18][19]
La estenosis pulmonar, o pulmonar, es un estrechamiento a menudo congénito de la válvula pulmonar; puede estar presente en los lactantes con afectación nasodigitoacústica.[4] El tratamiento de esta anomalía cardiaca puede requerir cirugía, o procedimientos no quirúrgicos como la valvuloplastia con balón (ensanchamiento de la válvula con un catéter de globo catéter).[20]
Historia
El síndrome fue descrito inicialmente en 1973 por James A. Keipert y colaboradores. Informaron de dos hermanos con falanges distales anchas, hipoacusia neurosensorial y rasgos faciales compatibles con lo que se conocería como síndrome de Keipert o "nasodigitoacústico".[4][2]
Epidemiología
Aunque no se ha determinado una tasa específica de incidencia, el síndrome está considerado una enfermedad rara tanto por la Oficina de Enfermedades Raras (ORDR) de los Institutos Nacionales de Salud, como por Orphanet. Esto sugiere, respectivamente, que el síndrome nasodigitoacústico afecta a menos de 200.000 personas en EE. UU., o que no afecta a más de 1 de cada 2.000 personas en Europa.[5]
Referencias
- ↑ OMIM 301026
- ↑ a b c d e f Keipert, JA; Fitzgerald, MG; Danks, DM (Feb 1973). "A new syndrome of broad terminal phalanges and facial abnormalities". Australian Paediatric Journal. 9 (1): 10–13. doi:10.1111/j.1440-1754.1973.tb02215.x. PMID 4708024. S2CID 30695579.
- ↑ a b c d e Amor, D. J.; Dahl, H-H.; Bahlo, M.; Bankier, A. (Oct 2007). "Keipert syndrome (Nasodigitoacoustic syndrome) is X-linked and maps to Xq22.2–Xq28". American Journal of Medical Genetics Part A. 143A (19): 2236–2241. doi:10.1002/ajmg.a.31917. PMID 17726694. S2CID 34320632.
- ↑ a b c d e f g h Cappon, SM; Khalifa, MM (Jul–Aug 2000). "Additional case of Keipert syndrome and review of the literature" (Free full text). Medical Science Monitor. 6 (4): 776–778. PMID 11208408.
- ↑ a b "Prevalence and Incidence of Nasodigitoacoustic syndrome". WrongDiagnosis.com. Consultado Abril 7, 2011.
- ↑ a b Balci, S; Dagli, S (Oct 1996). "Keipert syndrome in two brothers from Turkey". Clinical Genetics. 50 (4): 223–228. doi:10.1111/j.1399-0004.1996.tb02631.x. PMID 9001804. S2CID 34188534.
- ↑ a b c d e f Nik-Zainal, S.; Holder, S. E.; Cruwys, M.; Hall, C. M.; Shaw-Smith, C. (Jul 2008). "Keipert syndrome: two further cases and review of the literature". Clinical Dysmorphology. 17 (3): 169–175. doi:10.1097/MCD.0b013e3282f4afc3. PMID 18541962.
- ↑ a b Gorlin (1995). illustration, p. 209.
- ↑ a b Gorlin (1995). p. 208
- ↑ a b c Dumic, M.; Kokic, D. D.; Matic, T.; Potocki, K. (Nov 2006). "Daughter and her mildly affected father with Keipert syndrome". American Journal of Medical Genetics Part A. 140A (22): 2488–2492. doi:10.1002/ajmg.a.31489. PMID 17036315. S2CID 2978286.
- ↑ Reardon, W.; Hall, C. M. (Apr 2003). "Broad thumbs and halluces with deafness: A patient with Keipert syndrome". American Journal of Medical Genetics. 118A (1): 86–89. doi:10.1002/ajmg.a.10063. PMID 12605449. S2CID 27419345.
- ↑ Amor DJ, Stephenson SEM, Mustapha M, Mensah MA, Ockeloen CW, Lee WS, Tankard RM, Phelan DG, Shinawi M, de Brouwer APM, Pfundt R, Dowling C, Toler TL, Sutton VR, Agolini E, Rinelli M, Capolino R, Martinelli D, Zampino G, Dumić M, Reardon W, Shaw-Smith C, Leventer RJ, Delatycki MB, Kleefstra T7, Mundlos S, Mortier G, Bahlo M, Allen NJ, Lockhart P (2019) Pathogenic variants in GPC4 cause Keipert syndrome. Am J Hum Genet
- ↑ OMIM 245150
- ↑ OMIM 602849
- ↑ OMIM 184460
- ↑ OMIM 180849
- ↑ Feghali, J.; Lefebvre, P.; Staecker, H.; Kopke, R.; Frenz, D.; Malgrange, B.; Liu, W.; Moonen, G.; Ruben, R.; Van De Water, T. R. (Apr 1998). "Mammalian auditory hair cell regeneration/repair and protection: A review and future directions". Ear, Nose, & Throat Journal. 77 (4): 276, 280, 282–285. doi:10.1177/014556139807700409. PMID 9581394. S2CID 46494159.
- ↑ Smith, R.; Hildebrand, M.; Van Camp, G.; Pagon, R.; Bird, T.; Dolan, C.; Stephens, K. (1993). "Genetic Hearing Loss Overview". Deafness and Hereditary Hearing Loss Overview. University of Washington, Seattle. PMID 20301607.
- ↑ Kikidis, D.; Nikolopoulos, T. P.; Kampessis, G.; Stamatiou, G.; Chrysovergis, A. (2011). "Sudden Sensorineural Hearing Loss: Subclinical Viral and Toxoplasmosis Infections as Aetiology and How They Alter the Clinical Course". ORL. 73 (2): 110–115. doi:10.1159/000324210. PMID 21389742. S2CID 25767318.
- ↑ Ali Khan, M.; Al-Yousef, S.; Huhta, J.; Bricker, J.; Mullins, C.; Sawyer, W. (Mayo 1989). "Critical pulmonary valve stenosis in patients less than 1 year of age: Treatment with percutaneous gradational balloon pulmonary valvuloplasty". American Heart Journal. 117 (5): 1008–1014. doi:10.1016/0002-8703(89)90854-5. PMID 2711961.
Bibliografía
- Gorlin, R. J.; Toriello, H. V.; Cohen, M. M. (1995). Hereditary hearing loss and its syndromes. U. S.: Oxford University Press. pp. 208–209. ISBN 9780195065527. Recuperado el 21 de abril de 2011.