Síndrome del cuerno occipital
| El síndrome del cuervo occipital | ||
|---|---|---|
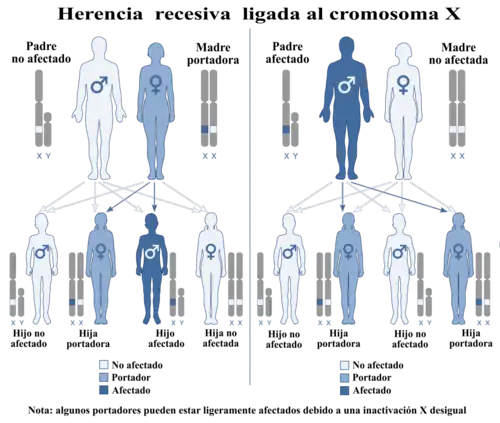 Esta enfermedad es heredara de tipo [recesiva ligada al cromosoma X]. | ||
| Especialidad | Endocrinología | |
| Complicaciones | Aneurisma de aorta | |
| Medicación | Droxidopa, inyecciones de histidina de cobre | |
| Sinónimos | ||
| Síndrome de Ehlers-Danilo’s tipo IX; Cutis Laxa Ligada al Cromosoma X | ||
El síndrome del cuerno occipital, antes considerado una variante del síndrome Ehlers-Danlos,[1] es un trastorno del tejido conectivo y enfermedad mitocondrial de tipo recesivo y ligado al cromosoma X. La causa de esta enfermedad es la deficiencia en el transporte de un mineral esencial, el cobre, y se asocia con mutaciones en el gen ATP7A.[2][3]
Aproximadamente, solo dos tercios de los niños con el síndrome del cuerno occipital son considerados como portadores del trastorno heredado genéticamente; el otro tercio no tiene un historial de la enfermedad en su familia. Esta enfermedad afecta a los hombres mayormente, ya que es un trastorno recesivo ligado al cromosoma X. Esto es debido a que los hombres no tienen un segundo cromosoma X como las mujeres y les falta esencialmente una segunda copia de respaldo que funcione correctamente. Las mujeres tienen más probabilidades de ser portadoras del trastorno. Una mujer con este trastorno tendría ambos cromosomas X afectados, no solo uno.[4]
Este trastorno es considerado una variante más leve del síndrome del cabello acerado. [5]
Signos y síntomas
El síndrome del cuerno occipital se caracteriza por la deficiencia de la excreción biliar de cobre, lo que causa deformidades en el esqueleto. Estos pueden incluir protuberancias en la parte trasera del cráneo (el hueso parasagital sobresale del hueso occipital, lo que se conoce como “cuernos occipitales”) y también deformidades en el codo, la dislocación de la cúpula radial, los extremos laterales de las clavículas en forma de martillo y anomalías en la cadera y la pelvis.[6] El síndrome del cuerno occipital puede presentarse a una edad temprana o más adelante a la niñez.[7] Los niños pueden tener los siguientes riesgos:
- Inteligencia normal o un leve retraso
- Cuello largo, paladar alto y arqueado en la boca, una cara larga y una frente alta
- Flacidez en la piel o hipermovilidad articular
- Hernia inguinal
- Retorcimiento de los vasos sanguíneos
- Divertículos de la vejiga
- Disautonomía (la incapacidad de regular partes del sistema nervioso)
- Diarrea crónica
- Vello grueso
- Niveles bajos de ceruloplasmina (9-29mg/dL)
[8]
Causa
El síndrome del cuerno occipital es una variante alélica más leve de la enfermedad de Menkes, la cual comienza a una edad más avanzada y está asociada con una neurodegeneración central menos severa. La naturaleza menos severa del síndrome del cuerno occipital suele atribuir a las mutaciones en las uniones, lo cual permite que hasta el 20 % a un 30 % de las transcripciones de ARN mensajero (ARNm) de ATP7A sean procesadas correctamente. Al igual que en los casos de la enfermedad de Menkes, las personas con el síndrome del cuerno occipital presentan anomalías en el tejido conjuntivo como resultado de la actividad deficiente de la lisil oxidasa, una enzima que requiere cobre y que usualmente desamina lisina e hydroxilisina en el primer paso de formación de entrecruzamientos de colágeno. Las personas también frecuentemente padecen de signos y síntomas de disautonomía relacionados con una deficiencia parcial en la actividad de la dopamina beta-hidroxilasa. La dopamina beta-hidroxilasa es otra enzima que depende del cobre y que usualmente convierte la dopamina en norepinefrina, un neurotransmisor crucial en las neuronas norepinefrinas. Un modelo natural de ratones con el síndrome del cuerno occipital, conocido como el modelo moteado, recapitula las anomalías del tejido cojuntivo, la deficiencia de la dopamina beta-hidroxilasa y el daño leve del sistema nervioso central en los humanos. [10]
Diagnóstico
El diagnóstico inicial de la enfermedad de Menkes y sus variantes más leves, como el síndrome del cuerno occipital, se basa en los síntomas clínicos. Los niveles bajos de cobre y ceruloplasmina sérica respaldan clínicamente la sospecha de la enfermedad del cuerno occipital, pero la confirmación bioquímica mediante cultivo de tejidos es necesaria. La evidencia del diagnóstico es la demostración del defecto molecular en ATP7A. La presencia de protuberancias en los huesos del occipucio da un diagnóstico definitivo, lo cual se observa en algunos pacientes.[11]
Tratamiento
El tratamiento para los niños con el síndrome del cuerno occipital depende de qué tan serio sea su caso. Los niños con OHS reciben frecuentemente terapia física y ocupacional.[12] Pueden requerir suplementos alimenticios mediante una sonda nasogástrica si no presentan un crecimiento adecuado. Con el objetivo de mejorar las manifestaciones neurológicas, como las convulsiones, el niño puede recibir inyecciones de histidina de cobre o colorido de cobre a una edad temprana. Sin embargo, las inyecciones de histidina de cobre han demostrado ser ineficaces para tratar con manifestaciones del tejido conectivo del OHS.[13]
Pronóstico
El historial de largo plazo de OHS es desconocido.[14] Algunos pacientes han fallecido a la edad joven de 17 años[15] mientras que otro paciente ha sobrevivido hasta los 57 años.[16] Las causas de muerte incluyen fallo respiratorio,[17] aneurisma aórtica[18] y hemorragia intracraneal.[19]
Investigaciones
Los Institutos Nacionales de Salud (NIH, por sus siglas en inglés) y Cyprium Therapeutics han coordinado el desarrollo de una terapia genética de un virus adenoasociado llamado AAV-ATP7A para la enfermedad Menkes y sus variantes, como el síndrome del cuerno occipital.[20] En marzo del 2017, Cyprium Therapeutics consiguió los derechos comerciales y el desarrollo mundial del programa Menkes con los Institutos Nacionales de la Salud y el Instituto Nacional de Salud Infantil y Desarrollo Humano a través de un acuerdo de cooperación y desarrollo de investigación (CRADA, por sus siglas en inglés) y acuerdos de licencia con el instituto Nacional de la Salud Infantil y Desarrollo Humano. [21] Aunque ha recibido una designación de medicamente huérfano por la Administración de Alimentos y Medicamentos (FDA, por sus siglas en inglés), el AAV-ATP7A permanece en etapa preclínica.[cita requerida]
Véase también
Referencias
- ↑ OMIM 304150
- ↑ Scheiber, Ivo; Dringen, Ralf; Mercer, Julian F. B. (2013). «Chapter 11. Copper: Effects of Deficiency and Overload». En Astrid Sigel, Helmut Sigel and Roland K. O. Sigel, ed. Interrelations between Essential Metal Ions and Human Diseases. Metal Ions in Life Sciences 13. Springer. pp. 359-387. ISBN 978-94-007-7499-5. PMID 24470097. doi:10.1007/978-94-007-7500-8_11.
- ↑ Tang J., Robertson S., Lem K.E., Godwin S.C., Kaler S.G. (November 2006). «Functional copper transport explains neurologic sparing in occipital horn syndrome». Genetics in Medicine 8 (11): 711-8. PMID 17108763. doi:10.1097/01.gim.0000245578.94312.1e.
- ↑ Horn Syndrome (enlace roto disponible en este archivo)., 9 August 2004.
- ↑ Kennerson ML, Nicholson GA, Kaler SG, Kowalski B, Mercer JF, Tang J, Llanos RM, Chu S, Takata RI, Speck-Martins CE, Baets J, Almeida-Souza L, Fischer D, Timmerman V, Taylor PE, Scherer SS, Ferguson TA, Bird TD, De Jonghe P, Feely SM, Shy ME, Garbern JY (March 2010). «Missense mutations in the copper transporter gene ATP7A cause X-linked distal hereditary motor neuropathy». American Journal of Human Genetics 86 (3): 343-52. PMC 2833394. PMID 20170900. doi:10.1016/j.ajhg.2010.01.027.
- ↑ OMIM 304150
- ↑ Horn Syndrome «Archive.org» (enlace roto disponible en este archivo)., 9 August 2004.
- ↑ Kodama H, Murata Y, Kobayashi M (August 1999). «Clinical manifestations and treatment of Menkes disease and its variants». Pediatrics International 41 (4): 423-9. PMID 10453199. S2CID 21267747. doi:10.1046/j.1442-200x.1999.01095.x.
- ↑ a b Wakai S, Ishikawa Y, Nagaoka M, Okabe M, Minami R, Hayakawa T (May 1993). «Central nervous system involvement and generalized muscular atrophy in occipital horn syndrome: Ehlers-Danlos type IX. A first Japanese case». Journal of the Neurological Sciences 116 (1): 1-5. PMID 8099605. S2CID 40538663. doi:10.1016/0022-510x(93)90081-9.
- ↑ Kaler SG (January 2011). «ATP7A-related copper transport diseases-emerging concepts and future trends». Nature Reviews. Neurology 7 (1): 15-29. PMC 4214867. PMID 21221114. doi:10.1038/nrneurol.2010.180.
- ↑ Horn, Nina; Tümer, Zeynep (2003). «Chapter 14: Menkes Disease and the Occipital Horn Syndrome». En Royce; Steinmann, eds. Connective Tissue and Its Heritable Disorders: Molecular, Genetic, and Medical Aspects (Second edición). Wiley-Blackwell. pp. 651-685. ISBN 978-0-471-25185-9. doi:10.1002/0471221929.ch14.
- ↑ Horn Syndrome (enlace roto disponible en este archivo)., 9 August 2004.
- ↑ Kodama H, Fujisawa C, Bhadhprasit W (March 2011). «Pathology, clinical features and treatments of congenital copper metabolic disorders--focus on neurologic aspects». Brain & Development 33 (3): 243-51. PMID 21112168. doi:10.1016/j.braindev.2010.10.021.
- ↑ «ATP7A-related copper transport diseases-emerging concepts and future trends». Nature Reviews. Neurology 7 (1): 15-29. January 2011. PMC 4214867. PMID 21221114. doi:10.1038/nrneurol.2010.180.
- ↑ «Occipital horn syndrome and classical Menkes Syndrome caused by deep intronic mutations, leading to the activation of ATP7A pseudo-exon». European Journal of Human Genetics 22 (4): 517-21. April 2014. PMC 3953917. PMID 24002164. doi:10.1038/ejhg.2013.191.
- ↑ «ATP7A pathogenic variant in a family exhibiting a variable occipital horn syndrome phenotype». Molecular Genetics and Metabolism Reports 13: 14-17. December 2017. PMC 5522958. PMID 28761814. doi:10.1016/j.ymgmr.2017.07.007.
- ↑ «Occipital horn syndrome and classical Menkes Syndrome caused by deep intronic mutations, leading to the activation of ATP7A pseudo-exon». European Journal of Human Genetics 22 (4): 517-21. April 2014. PMC 3953917. PMID 24002164. doi:10.1038/ejhg.2013.191.
- ↑ Quiroga, Elina; Heneghan, Rachel (June 2015). «Abdominal aortic aneurysm in a patient with occipital horn syndrome 2». Journal of Vascular Surgery Cases 1 (2): 138-140. PMC 6849971. PMID 31725130. doi:10.1016/j.jvsc.2015.03.012.
- ↑ «Distinctive Menkes disease variant with occipital horns: delineation of natural history and clinical phenotype». American Journal of Medical Genetics 65 (1): 44-51. October 1996. PMID 8914740. doi:10.1002/(SICI)1096-8628(19961002)65:1<44::AID-AJMG7>3.0.CO;2-Y.
- ↑ «ATP7A gene addition to the choroid plexus results in long-term rescue of the lethal copper transport defect in a Menkes disease mouse model». Molecular Therapy 19 (12): 2114-23. December 2011. PMC 3242653. PMID 21878905. doi:10.1038/mt.2011.143.
- ↑ «Corporate Presentation». Cyprium Therapeutics, Inc. October 2017.