Síndrome de hendidura facial de Malpuech
| Síndrome de hendidura facial de Malpuech | ||
|---|---|---|
| Especialidad | Genética médica | |
El síndrome de hendidura facial de Malpuech, también llamado síndrome de Malpuech o síndrome de hendidura facial de tipo gitano,[1] es un síndrome congénito poco frecuente. Se caracteriza por hendidura facial (cualquier tipo de hendidura en los huesos y tejidos de la cara, incluidos labio leporino y paladar hendido), un apéndice caudal (una «cola humana»),[2][3] deficiencia del crecimiento, discapacidad intelectual y del desarrollo, y anomalías del sistema renal (riñones) y de los genitales masculinos.[4] También puede haber anomalías cardíacas y otras malformaciones esqueléticas.[5] El síndrome fue descrito inicialmente por Georges Malpuech y colaboradores en 1983.[6] Se cree que está relacionado genéticamente con el síndrome de Juberg-Hayward. El síndrome de Malpuech también se ha considerado parte de un espectro de trastornos genéticos congénitos asociados a anomalías faciales, urogenitales y esqueléticas similares. Denominado «síndrome 3MC», este espectro propuesto incluye los síndromes de Malpuech, Michels y Mingarelli-Carnevale (OSA).[7][8] Se cree que las mutaciones en los genes COLLEC11 y MASP1 son la causa de estos síndromes.[9] Se desconoce la incidencia del síndrome de Malpuech. El patrón de herencia es autosómico recesivo, lo que significa que un gen defectuoso (mutado) asociado con el síndrome se localiza en un autosoma, y el síndrome se produce cuando se heredan dos copias de este gen defectuoso.[10]
Señales y síntomas
El síndrome de Malpuech es congénito y se manifiesta al nacer. Se caracteriza por un rasgo conocido como hendidura facial. Observada y señalada en

la descripción inicial del síndrome como labio leporino y paladar hendido,[6] la hendidura facial se identifica por hendiduras en los huesos, músculos y tejidos de la cara, incluidos los labios y el paladar. Las formas de labio leporino y paladar hendido que se observan típicamente con el síndrome de Malpuech son la línea media (por la mitad del labio y el paladar)[11] o bilateral (que afecta a ambos lados de la boca y el paladar).[10] Las hendiduras faciales generalmente abarcan una amplia gama de gravedad, que van desde anomalías menores como una úvula bífida (partida), labio leporino y paladar hendido, hasta importantes defectos estructurales y de desarrollo de los huesos faciales y los tejidos blandos.[12] La hendidura del labio y el paladar se produce durante la embriogénesis.[13][14] Otras anomalías faciales y ortodentales que se han descrito con el síndrome incluyen: hipertelorismo (ojos inusualmente separados, a veces denominado telecanto), fisuras palpebrales estrechas (la separación entre los párpados superior e inferior) y ptosis (caída) de los párpados, protuberancia frontal (cresta de la ceja prominente) con sinofris, cejas muy arqueadas, raíz nasal ancha y punta nasal aplanada, hipoplasia malar (pómulo superior subdesarrollado), micrognatia (mandíbula inferior demasiado pequeña) e incisivos prominentes. Las anomalías auditivas incluyen un reborde de la oreja agrandado y deficiencias auditivas asociadas a otitis media congénita (u «oído de pegamento», inflamación del oído medio) y pérdida de audición neurosensorial.[4][15][16]
Otra característica identificada con el síndrome de Malpuech es un apéndice caudal.[1][3] Un apéndice caudal es una excrecencia congénita que sale del cóccix (rabadilla). Presente en muchas especies animales no humanas como una cola típica, esta característica cuando se observa en un bebé se ha descrito como una «cola humana».[2][17] Esto fue observado por Guion-Almeida (1995) en tres individuos de Brasil. En las radiografías, el apéndice aparecía como una protuberancia prominente del cóccix.[3] En el examen físico, el apéndice se parece a un nódulo en forma de cola de animal.[18]
Son frecuentes deficiencias como discapacidad intelectual, problemas de aprendizaje, retraso del crecimiento y retraso del desarrollo. Las manifestaciones psiquiátricas que se han descrito con el síndrome incluyen comportamiento psicótico, trastorno obsesivo-compulsivo, pérdida de inhibición, hiperactividad, agresividad, miedo al contacto físico y acciones compulsivas como la ecolalia (repetir las palabras pronunciadas por otra persona). También se han observado tics neuromusculares.[4][15]
Las anomalías urogenitales, o las que afectan a los sistemas urinario y reproductor, son frecuentes en el síndrome. Malpuech et al. (1983) y Kerstjens-Frederikse et al. (2005) informaron de varias formas en varones afectados: micropene, hipospadias (una mala localización congénita del meato urinario), criptorquidia (testículos ectópicos o no descendidos), escroto bífido (partido) y subdesarrollado, y una válvula uretral obstructiva.[4][6] Reardon et al. (2001) también informaron de un niño afectado con agenesia renal izquierda, riñón derecho agrandado y desplazado hacia abajo, criptorquidia y escroto en chal.[7] Otras malformaciones que se han observado con el síndrome son onfalocele[3] y hernia umbilical.[19]
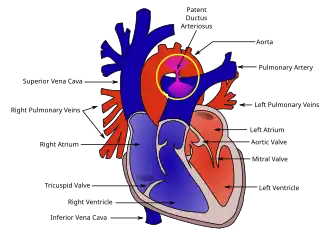
También se han observado anomalías congénitas del corazón en el síndrome de Malpuech. A partir de una pareja japonesa sana, Chinen y Naritomi (1995) describieron al sexto niño que presentaba características compatibles con el trastorno. Este lactante varón de dos meses de edad también estaba afectado por anomalías cardiacas que incluían conducto arterioso persistente (CAP)[20] y defecto septal ventricular.[5] La abertura en el conducto arterioso asociada al CAP había sido reparada quirúrgicamente en el lactante a los 38 días de edad. También se informó de una serie de aberraciones esqueléticas menores en el lactante, incluidos huesos wormianos en las suturas lambdoideas.[5]
Genética
El síndrome de Malpuech, al igual que el resto de trastornos dentro de la consideración de síndrome 3MC, está causado por mutaciones en los genes COLLEC11 y MASP1. En una investigación de Rooryck et al. (2011), se estudiaron once familias afectadas por el síndrome 3MC, lo que dio lugar a la identificación de estas dos mutaciones. Ambos genes codifican proteínas de la vía del complemento lectina, que desempeña un papel en el sistema del complemento de la inmunidad innata, o inespecífica, en humanos y otras especies.[9]
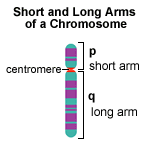
El gen COLLEC11 o CL-K1 está localizado en el brazo corto del cromosoma 2 (2p25.3) en humanos.[21] La proteína CL-K1 es una lectina de tipo C y pertenece a la familia de las colectinas. Además de su papel en la inmunidad innata, se cree que la proteína interviene en el desarrollo de tejidos como el cartílago craneofacial, el corazón y el riñón durante la embriogénesis. Esta función en el desarrollo facial se corroboró mediante el estudio del pez cebra, en el que mutaciones en su versión de CL-K1 contribuyeron a anomalías craneofaciales (como las hendiduras craneofaciales) posiblemente asociadas a errores en la migración de las células de la cresta neural.[9]
El gen MASP1, o Proteasa I de Serina de unión a Manano, está situado en el brazo largo del cromosoma humano 3 en 3q27-q28.[22] La proteína es un tipo de conectina llamada lectina de unión a manosa, que desempeña un papel en la inmunidad innata al unirse a patógenos como los virus, incluido el VIH.[23]
Según lo descrito por Sirmaci et al. (2010), se evaluaron tres individuos turcos de dos familias consanguíneas (se dice que los hijos de parientes como primos pertenecen a una familia consanguínea) con diversas características del síndrome 3MC, incluido el dismorfismo facial y un apéndice caudal. La investigación de cromosomas homólogos mediante mapeo genético reveló una región autocigótica (una localización en un cromosoma donde ambos alelos de un gen proceden de un ancestro común) en el cromosoma 3q27 en ambas familias. En una familia, una mutación sin sentido en MASP1 en esta localización provocó la sustitución del aminoácido glicina por arginina en la posición 687 de la secuencia genética. La mutación se corregía con el fenotipo observado. En los individuos de la segunda familia, la secuenciación del ADN de MASP1 mostró una mutación sin sentido que resultó en una desactivación del triptófano en la posición 290 del gen, que también se cosegregó con el fenotipo. Ambas mutaciones se producen en una forma de MASP1 conocida por procesar IGFBP5; la pérdida de esta función asociada a la mutación de MASP1 provoca alteraciones en la disponibilidad del factor de crecimiento similar a la insulina durante el desarrollo craneofacial y musculoesquelético durante el periodo embrionario. Estos resultados indican que las mutaciones en MASP1 son responsables de una serie de características que se encuentran en los trastornos por malformaciones, incluido el síndrome de Malpuech.[24]
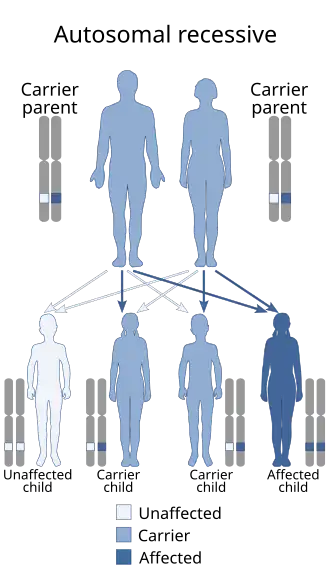
El síndrome se hereda de forma autosómica recesiva,[10] lo que significa que el gen o genes defectuosos responsables del trastorno (COLLEC11, MASP1) se localizan en un autosoma (los cromosomas 2 y 3 son autosomas), y se necesitan dos copias del gen defectuoso (una heredada de cada progenitor) para nacer con el trastorno. Los padres de un individuo con un trastorno autosómico recesivo son ambos portadores de una copia del gen defectuoso, pero normalmente no experimentan ningún signo o síntoma del trastorno.
Diagnóstico
Se sugiere que los criterios de diagnóstico del síndrome de Malpuech incluyan labio leporino y/o paladar hendido, rasgos faciales típicos asociados y al menos dos de los siguientes: anomalías urogenitales, apéndice caudal y retraso del crecimiento o del desarrollo.[25] Debido a la tasa relativamente alta de discapacidad auditiva que se encuentra en este trastorno, también puede tenerse en cuenta en el diagnóstico. Otro trastorno congénito, el síndrome de Wolf-Hirschhorn (Pitt-Rogers-Danks), comparte las características de Malpuech en sus criterios diagnósticos. Debido a esta falta de diferenciación, puede emplearse el cariotipo (análisis microscópico de los cromosomas de un individuo) para distinguir ambos. Mientras que las deleciones en el brazo corto del cromosoma 4 se revelarían con Wolf-Hirschhorn, un cariotipo sin esta aberración presente favorecería un diagnóstico de síndrome de Malpuech. Además, el cariotipo de un individuo con síndrome de Malpuech solo será normal.[15]
Clasificación
Se ha demostrado que el síndrome de Malpuech presenta similitudes físicas o fenotípicas con varios otros trastornos genéticos. Un informe de Reardon et al. (2001) de un niño de nueve años que presentaba anomalías faciales, caudales y urogenitales compatibles con el síndrome de Malpuech, que también tenía malformaciones esqueléticas indicativas del síndrome de Juberg-Hayward, sugiere que los dos trastornos pueden ser alélicos (causados por diferentes mutaciones del mismo gen).[7]
Junto con otros trastornos que tienen características similares o superpuestas y herencia autosómica recesiva, se ha considerado que el síndrome de Malpuech pertenece a la denominación «síndrome 3MC». Titomanlio et al. (2005) describieron a una niña de tres años con síndrome de Michels. En su revisión de las similitudes físicas entre los síndromes de Michels, Malpuech y Mingarelli-Carnevale -particularmente la apariencia facial, incluyendo casos de labio leporino y paladar hendido, y ptosis, y una similitud de anomalías congénitas abdominales y urogenitales- creían que los síndromes podrían representar un espectro de trastornos genéticos en lugar de tres trastornos individuales. Inicialmente sugirieron que este espectro podría denominarse síndrome 3MC (Michels-Malpuech-Mingarelli-Carnevale).[8] Esta conclusión y el nombre de síndrome 3MC fueron respaldados por Leal et al. (2008), que informaron de un hermano y una hermana con una serie de síntomas que se solapaban con los distintos síndromes.[26] Rooryck et al. (2011) siguieron afirmando el síndrome 3MC al explicar su causa.[9]
Tratamiento
Muchas de las malformaciones congénitas del síndrome de Malpuech pueden corregirse quirúrgicamente. Entre ellas se incluyen labio leporino y paladar hendido, onfalocele, anomalías urogenitales y craneofaciales, deformidades esqueléticas como apéndice caudal o escoliosis, y hernias del ombligo. El principal motivo de preocupación en estos procedimientos aplicados a un neonato con trastornos congénitos, incluido el síndrome de Malpuech, es la logística de la anestesia. Métodos como la intubación traqueal para el manejo de las vías respiratorias durante la anestesia general pueden verse dificultados por la boca aún más pequeña o mal desarrollada del lactante. Para la anestesia regional, métodos como el bloqueo espinal son más difíciles cuando hay escoliosis. En un informe de 2010 de Kiernan et al., una niña de cuatro años con síndrome de Malpuech estaba siendo preparada para una amigdalectomía y adenoidectomía no relacionadas. Durante la intubación, la inserción de un laringoscopio, necesario para identificar las vías respiratorias para la colocación del tubo endotraqueal, resultó problemática por la presencia de micrognatia atribuida al síndrome. Tras sustituirlo por un laringoscopio de tamaño ajustado, la intubación transcurrió con normalidad. La anestesia general fue un éxito.[16]
Priolo et al. (2007) presentaron un seguimiento poco frecuente de un varón con síndrome de Malpuech. Nacido a término tras un embarazo y un parto sin complicaciones, el niño fue sometido a una reparación quirúrgica del labio leporino y el paladar hendido. No se informó de ningún problema con el procedimiento. También se detectó una anomalía cardiaca, una comunicación interauricular, que no requirió intervención. A los tres años se le diagnosticaron discapacidad intelectual, hiperactividad y trastorno obsesivo compulsivo; a los seis años se le diagnosticó una discapacidad auditiva, que se trató con audífonos. A lo largo de la década siguiente se realizaron varias evaluaciones psiquiátricas. A los 14 años mostraba miedo al contacto físico; a los 15 sufrió un episodio psicótico grave, caracterizado por agitación y pérdida de la inhibición sociosexual. Este conjunto de síntomas fue tratado farmacológicamente (con medicamentos de prescripción). A los 17 años mantenía un bajo nivel de deficiencia mental, con momentos de ecolalia compulsiva.[15]
Historia
No se ha determinado la incidencia del síndrome de Malpuech.[16] Un informe de Crisponi et al. de 1999 sugería que sólo unos 12 individuos en todo el mundo estaban afectados por el trastorno en ese momento.[19] El síndrome fue notificado por primera vez por Guilliaume Malpuech y sus colegas en 1983, observado en cuatro niños de sexo no especificado en lo que se describió como una familia gitana. Entre los niños había tres hermanos y un primo hermano; se sabía que la familia era muy consanguínea.[6][15]
Referencias
- ↑ a b «Entry - #248340 - 3MC SYNDROME 3; 3MC3 - OMIM - (OMIM.ORG)». omim.org (en inglés estadounidense). Consultado el 23 de junio de 2025.
- ↑ a b Adeleye, A. O.; Olowookere, K. G. (2010). «"A Human Tail in an Infant (Letter)"». West African Journal of Medicine. PMID 21089026.
- ↑ a b c d Guion-Almeida, M. L. (1995). «"Apparent Malpuech syndrome: Report on three Brazilian patients with additional signs"». American Journal of Medical Genetics. PMID 7573149. doi:10.1002/ajmg.1320580104.
- ↑ a b c d Kerstjens-Frederikse, W. S.; Brunner, H. G.; Van Dael, C. M.; Van Essen, A. J. (2005). «"Malpuech syndrome: Three patients and a review"». American Journal of Medical Genetics Part A. PMID 15793834. doi:10.1002/ajmg.a.30662.
- ↑ a b c Chinen, Yasutsugu; Naritomi, Kenji (1995-12). «Malpuech facial clefting syndrome in a Japanese boy with cardiac defects». Japanese journal of human genetics (en inglés) 40 (4): 335-338. ISSN 1435-232X. doi:10.1007/BF01900601. Consultado el 24 de junio de 2025.
- ↑ a b c d Malpuech, G.; Demeocq, F.; Palcoux, J. B.; Vanlieferinghen, P.; Opitz, J. M. (1983). «"A previously undescribed autosomal recessive multiple congenital anomalies/mental retardation (MCA/MR) syndrome with growth failure, lip/palate cleft(s), and urogenital anomalies".». American Journal of Medical Genetics. PMID 6660246. doi:10.1002/ajmg.1320160405.
- ↑ a b c Reardon, W.; Hall, C. M.; Gorman, W. (2001). «"An atypical case suggesting the possibility of overlap between Malpuech and Juberg-Hayward syndromes"». Clinical Dysmorphology. PMID 11310992. doi:10.1097/00019605-200104000-00009.
- ↑ a b Titomanlio, Luigi; Bennaceur, Selim; Bremond-Gignac, Dominique; Baumann, Clarisse; Dupuy, Olivier; Verloes, Alain (2005). «Michels syndrome, Carnevale syndrome, OSA syndrome, and Malpuech syndrome: Variable expression of a single disorder (3MC syndrome)?». American Journal of Medical Genetics Part A (en inglés). 137A (3): 332-335. ISSN 1552-4833. doi:10.1002/ajmg.a.30878. Consultado el 24 de junio de 2025.
- ↑ a b c d Rooryck, Caroline; Diaz-Font, Anna; Osborn, Daniel P. S.; Chabchoub, Elyes; Hernandez-Hernandez, Victor; Shamseldin, Hanan; Kenny, Joanna; Waters, Aoife et al. (2011-03). «Mutations in lectin complement pathway genes COLEC11 and MASP1 cause 3MC syndrome». Nature Genetics 43 (3): 197-203. ISSN 1546-1718. PMC 3045628. PMID 21258343. doi:10.1038/ng.757. Consultado el 24 de junio de 2025.
- ↑ a b c Turnbull, C.; Lees, M.; Chitty, L. S. (2006). «"Prenatal sonographic diagnosis of Malpuech syndrome".». Prenatal Diagnosis. PMID 17019743. doi:10.1002/pd.1564.
- ↑ «Healthgrades Health Library». Healthgrades (en inglés). Consultado el 24 de junio de 2025.
- ↑ «FACE - DIAGNOSIS OF CONGENITAL ABNORMALITIES - THE 18-23 WEEKS SCAN». centrus.com.br. Archivado desde el original el 21 de noviembre de 2010. Consultado el 24 de junio de 2025.
- ↑ «Facial Cleft -- Medical Definition». www.medilexicon.com (en inglés). Archivado desde el original el 31 de julio de 2016. Consultado el 24 de junio de 2025.
- ↑ «Rare facial cleft: 14-mth-old Hunan boy Kang Kang born with a 'mask' - What's On Xiamen». www.whatsonxiamen.com. Archivado desde el original el 12 de noviembre de 2018. Consultado el 24 de junio de 2025.
- ↑ a b c d e Priolo, M; Ciccone, R; Bova, I; Campolo, G; Lagana, C; Zuffardi, O (2007). «"Malpuech syndrome: Broadening the clinical spectrum and molecular analysis by array-CGH"». European Journal of Medical Genetics. PMID 17140870. doi:10.1016/j.ejmg.2006.10.004.
- ↑ a b c Kiernan, F; Crowe, S (2010). «"Malpuech syndrome: implications for anesthetic management".». Pediatric Anesthesia. PMID 20470345. doi:10.1111/j.1460-9592.2010.03271.x.
- ↑ Singh, D.; Kumar, B.; Sinha, V.; Bagaria, H. (2008). «"The human tail: rare lesion with occult spinal dysraphism—a case report".». Journal of Pediatric Surgery. PMID 18778987. doi:10.1016/j.jpedsurg.2008.04.030.
- ↑ Finn y Lynch (2006), ilustración, p. 243.
- ↑ a b Crisponi, G.; Marras, A. R.; Corrias, A. (1999). «"Two sibs with Malpuech syndrome".». American Journal of Medical Genetics. PMID 10482884. doi:10.1002/(SICI)1096-8628(19990917)86:3<294::AID-AJMG20>3.0.CO;2-2.
- ↑ «Entry - %607411 - PATENT DUCTUS ARTERIOSUS 1; PDA1 - OMIM - (OMIM.ORG)». omim.org (en inglés estadounidense). Consultado el 24 de junio de 2025.
- ↑ «Entry - *612502 - COLLECTIN 11; COLEC11 - OMIM - (OMIM.ORG)». omim.org (en inglés estadounidense). Consultado el 24 de junio de 2025.
- ↑ «Entry - *600521 - MANNAN-BINDING LECTIN SERINE PROTEASE 1; MASP1 - OMIM - (OMIM.ORG)». omim.org (en inglés estadounidense). Consultado el 24 de junio de 2025.
- ↑ Ji, X; Gewurz, H; Spear, G (2005). «"Mannose binding lectin (MBL) and HIV"». Molecular Immunology. PMID 15488604. doi:10.1016/j.molimm.2004.06.015.
- ↑ Sirmaci, Asli; Walsh, Tom; Akay, Hatice; Spiliopoulos, Michail; Sakalar, Yıldırım Bayezit; Hasanefendioğlu-Bayrak, Aylin; Duman, Duygu; Farooq, Amjad et al. (12 de noviembre de 2010). «MASP1 mutations in patients with facial, umbilical, coccygeal, and auditory findings of Carnevale, Malpuech, OSA, and Michels syndromes». American Journal of Human Genetics 87 (5): 679-686. ISSN 1537-6605. PMC 2978960. PMID 21035106. doi:10.1016/j.ajhg.2010.09.018. Consultado el 24 de junio de 2025.
- ↑ Finn, S. M.; Lynch, S. A. (2006). «"Malpuech syndrome: facial features in the absence of clefting".». Clinical Dysmorphology. PMID 16957483. doi:10.1097/01.mcd.0000220621.85896.46.
- ↑ Leal, G. F.; Silva, E. O.; Duarte, A. R.; Campos, J. F. (2008). «Blepharophimosis, blepharoptosis, defects of the anterior chamber of the eye, caudal appendage, radioulnar synostosis, hearing loss and umbilical anomalies in sibs: 3MC syndrome?"». American Journal of Medical Genetics Part A. PMID 18266249. doi:10.1002/ajmg.a.32252.