Síndrome de Omenn
| Síndrome de Omenn | ||
|---|---|---|
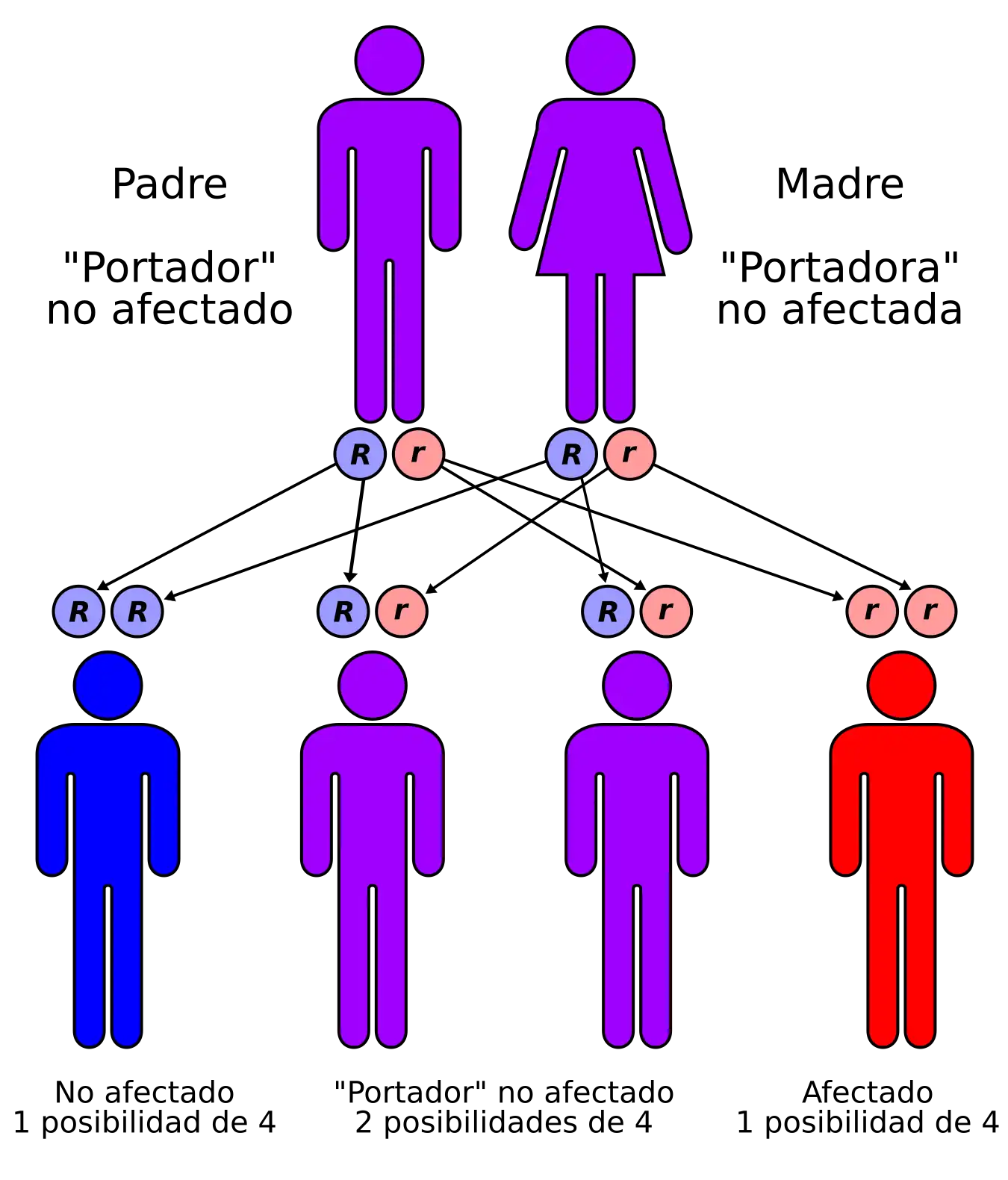 El síndrome de Omenn tiene un patrón hereditario autosómico recesivo. | ||
| Especialidad | hematología | |
| Sinónimos | ||
|
Reticuloendoteliosis familiar con eosinofilia Inmunodeficiencia combinada con hipereosinofilia | ||
El Síndrome de Omenn o reticuloendoteliosis familiar con eosinofilia es una inmunodeficiencia combinada severa autosómico recesiva[1] asociada con mutaciones con cambio de sentido en los genes activadores de recombinación (RAG1 y RAG2), que afectan a los niveles circulantes de los linfocitos B y T.
Sintomatología y Causas
Los síntomas son muy similares a la enfermedad del implante contra el hospedador (GVHD). Esto sucede porque los pacientes tienen algunos linfocitos T con niveles limitados de recombinación debido a que la función de los genes RAG son pobres. Estos linfocitos T son anormales y tienen una elevada especificidad para autoantígenos del timo y su periferia. Por tanto, estos linfocitos son autorreactivos y provocan el fenotipo GVHD.
Entre los síntomas se encuentran:
- Descamación
- Diarrea crónica.
- Eritrodermia (Enrojecimiento extenso de la piel)
- Hepatosplenomegalia (agrandamiento simultáneo del hígado y el bazo)
- Leucocitosis (Elevación de la cuenta de leucocitos)
- Linfadenopatía (Hinchazón de uno o más ganglios linfáticos)
- infecciones bacterianas de repetición
- Inmunoglobulina E (IgE) sérica elevada
Tratamiento
Inicialmente tratamiento inmunosupresor (corticoides, ciclosporina) para tratar la inflamación seguido de trasplante de médula ósea.[2]
Referencias
- ↑ Santagata S, Villa A, Sobacchi C, Cortes P, Vezzoni P (2000). «The genetic and biochemical basis of Omenn syndrome». Immunol Rev. 178: 64-74. PMID 11213808. doi:10.1034/j.1600-065X.2000.17818.x.
- ↑ «Continuum: Inmunodeficiencia combinada grave: avances recientes y orientación para su manejo clínico». continuum.aeped.es. Consultado el 13 de mayo de 2025.
Enlaces externos
| Control de autoridades |
|
|---|
 Datos: Q2214419
Datos: Q2214419 Multimedia: Omenn syndrome / Q2214419
Multimedia: Omenn syndrome / Q2214419