Metabarcoding (español)
El metabarcoding es la codificación mediante barras del ADN/ARN (o eADN/eARN) de una manera que permite la identificación simultánea de muchos taxones dentro de la misma muestra. La principal diferencia es que el barcoding se centra en un organismo específico, mientras que el metabarcoding tiene como objetivo determinar la composición de especies dentro de una muestra.
Introducción

La técnica del metabarcoding consiste en una región génica variable corta, que es útil para la asignación taxonómica por regiones génicas altamente conservadas, que se pueden utilizar para el diseño de cebadores.[1] Esta idea del metabarcoding en general se originó en 2003 de investigadores de la Universidad de Guelph.[2]
El procedimiento de metabarcoding, al igual que el barcoding general, procede en orden a través de etapas de extracción de ADN, amplificación por PCR, secuenciación y análisis de datos. Se utilizan diferentes genes dependiendo de si el objetivo es codificar con barras una sola especie o metabarcoding varias especies. En este último caso, se utiliza un gen más universal. La codificación metabólica no utiliza ADN/ARN de una sola especie como punto de partida, sino ADN/ARN de varios organismos diferentes derivados de una muestra ambiental o masiva.
ADN Ambiental
El ADN ambiental o eDNA describe el material genético presente en muestras ambientales como sedimentos, agua y aire, incluyendo células enteras, ADN extracelular y organismos potencialmente completos.[3][4] El eDNA se puede tomar de muestras ambientales y preservadas, extraídas, amplificadas, secuenciadas y categorizarse en base a su secuencia.[5]A partir de esta información, es posible la detección y clasificación de especies. El eDNA puede provenir de piel, mucosas, saliva, esperma, secreciones, huevos, heces, orina, sangre, raíces, hojas, frutos, polen y cuerpos en descomposición de organismos más grandes, mientras que los microorganismos se pueden obtener completamente. [6][7][4]La producción de eDNA depende de la biomasa, edad y actividad de alimentación del organismo, así como de la fisiología, ciclo de vidal y uso del espacio.[4][8][9][10] Para 2019, los métodos en la investigación de eDNA se habían ampliado para poder evaluar comunidades enteras a partir de una sola muestra. Este proceso implica el metabarcoding, que se puede definir con precisión como el uso de cebadores de reacción en cadena de la polimerasa (PCR) generales o universales en muestras de ADN mixtas de cualquier origen, seguido de una secuenciación de próxima generación (NGS) de alto rendimiento para determinar la composición de especies de la muestra. Este método ha sido común en microbiología durante años, pero, a partir de 2020, recién está encontrando su lugar en la evaluación de macroorganismos. Las aplicaciones de metabarcoding de eDNA en todo el ecosistema tienen el potencial no solo de describir comunidades y biodiversidad, sino también de detectar interacciones y ecología funcional en grandes escalas espaciales, aunque puede estar limitado por lecturas falsas debido a contaminación u otros errores.[7][11][12][13]
En conjunto, el metabarcoding de eDNA aumenta la velocidad, la precisión y la identificación con respecto al metabarcoding tradicional y reduce el costo, pero necesita ser estandarizado y ser unificado, integrando la taxonomía y los métodos moleculares para un estudio ecológico completo. [14][15][16][17][18][19]
El metabarcoding de ADN ambiental tiene aplicaciones en el monitoreo de la diversidad en todos los hábitats y grupos taxonómicos, la reconstrucción de ecosistemas antiguos, las interacciones entre plantas y polinizadores, el análisis de la dieta, la detección de especies invasoras, las respuestas a la contaminación y el monitoreo de la calidad del aire. El metabarcoding de ADN ambiental es un método único que aún se encuentra en desarrollo y probablemente seguirá en constante cambio durante algún tiempo a medida que avance la tecnología y se estandaricen varios procedimientos. Sin embargo, a medida que se optimice y su uso se generalice, es probable que se convierta en una herramienta esencial para el monitoreo ecológico y el estudio de la conservación global. [19]
ADN comunitario
Desde el inicio de la secuenciación de alto rendimiento (HTS),[20] el uso del metabarcoding como herramienta de detección en biodiversidad ha generado un inmenso interés. [12] Sin embargo, aún no está claro con respecto a qué material de origen se utiliza para realizar análisis de metabarcoding (por ejemplo, ADN ambiental versus ADN comunitario). Sin claridad entre estos dos materiales de origen, las diferencias en el muestreo, así como las diferencias en los procedimientos de laboratorio, pueden afectar los procesos bioinformáticos posteriores utilizados para el procesamiento de datos y complicar la interpretación de los patrones espaciales y temporales de la biodiversidad. Aquí, buscamos diferenciar claramente entre los materiales de origen predominantes utilizados y su efecto en el análisis e interpretación posteriores para el metabarcoding de ADN ambiental de animales y plantas en comparación con el metabarcoding de ADN comunitario.[21]
Con el metabarcoding de ADN comunitario en animales y plantas, los datos de los diversos grupos objetivo se recolectan con mayor frecuencia en masa (por ejemplo, suelo, trampa de malestar o red), los individuos se retiran de otros restos de muestra y se agrupan antes de la extracción de ADN masiva. [12] Por el contrario, el eADN de macroorganismos se aísla directamente de un material ambiental (p. ej., suelo o agua) sin segregación previa de organismos individuales o material vegetal de la muestra y supone implícitamente que el organismo completo no está presente en la muestra. Por supuesto, las muestras de ADN comunitario pueden contener ADN de partes de tejidos, células y orgánulos de otros organismos (p. ej., contenido intestinal, ADN intracelular o extracelular cutáneo). Del mismo modo, las muestras de eADN de macroorganismos pueden capturar inadvertidamente organismos microscópicos completos que no son el objetivo (p. ej., protistas, bacterias). Por lo tanto, la distinción puede fallar al menos parcialmente en la práctica. [22]
Otra distinción importante entre el ADN comunitario y el eADN de macroorganismos es que las secuencias generadas a partir del metabarcoding metabólico del ADN comunitario se pueden verificar taxonómicamente cuando los especímenes no se destruyen en el proceso de extracción. En este caso, las secuencias se pueden generar a partir de especímenes de referencia utilizando la secuenciación de Sanger. Como las muestras para la codificación metabólica de ADN ambiental carecen de organismos completos, no se pueden hacer comparaciones in situ. Por lo tanto, las afinidades taxonómicas solo se pueden establecer comparando directamente las secuencias obtenidas (o mediante unidades taxonómicas operativas (MOTU) generadas bioinformáticamente) con secuencias anotadas taxonómicamente, como la base de datos de nucleótidos GenBank del NCBI,[23] BOLD,[24] o con bases de datos de referencia autogeneradas a partir de ADN secuenciado por Sanger.[25][26][27](La unidad taxonómica operativa molecular (MOTU) es un grupo identificado mediante el uso de algoritmos de agrupamiento y un porcentaje predefinido de similitud de secuencia, por ejemplo, 97%)).[28][22] Luego, para corroborar al menos parcialmente la lista resultante de taxones, se realizan comparaciones con métodos de estudio convencionales físicos, acústicos o visuales realizados al mismo tiempo o en comparación con registros históricos de estudios para una ubicación. [22]
Por lo tanto, la diferencia en el material de origen entre el ADN comunitario y el eADN tiene ramificaciones distintas para interpretar la escala de inferencia para el tiempo y el espacio sobre la biodiversidad detectada. A partir del ADN comunitario, está claro que las especies individuales se encontraron en ese tiempo y lugar, pero en el caso del eADN, el organismo que produjo el ADN puede estar río arriba del lugar muestreado,[29] o el ADN puede haber sido transportado en las heces de una especie depredadora más móvil (por ejemplo, aves que depositan eADN de peces, [30] o estaba presente previamente, pero ya no está activo en la comunidad y la detección se realiza a partir del ADN que se desprendió años o décadas antes.[31] Esto último significa que la escala de inferencia tanto en el espacio como en el tiempo debe considerarse cuidadosamente al inferir la presencia de la especie en la comunidad basándose en el eADN. [22]
Etapas del Metabarcoding
Hay seis etapas o pasos en el ADN barcoding y el metabarcoding.
Primero, se eligen regiones adecuadas de ADN barcoding para responder a una pregunta de investigación específica. La región de ADN barcode más comúnmente utilizada para animales es un segmento de aproximadamente 600 pares de bases del gen mitocondrial citocromo oxidasa I (CO1).[2] Este locus proporciona una gran variación de secuencia entre especies y, al mismo tiempo, una variación relativamente pequeña dentro de la misma especie.[32] Otras regiones de ADN barcode comúnmente utilizadas para la identificación de especies animales son las regiones de ADN ribosómico (ADNr), como 16S, 18S y 12S, y las regiones mitocondriales, como el citocromo B.[33][34][35][36] Estos marcadores tienen ventajas y desventajas y se usan para diferentes propósitos.[37][38] Por lo general, se necesitan regiones de ADN barcode más largas (de al menos 600 pares de bases) para una delimitación precisa de especies, especialmente para diferenciar parientes cercanos. La identificación del productor de restos de organismos, como heces, pelos y saliva, puede utilizarse como una medida indirecta para verificar la presencia o ausencia de una especie en un ecosistema. El ADN en estos restos suele ser de baja calidad y cantidad, por lo que en estos casos se utilizan códigos de barras más cortos de alrededor de 100 pares de bases. Del mismo modo, los restos de ADN en las heces suelen estar degradados, por lo que se necesitan ADN barcodes cortos para identificar las presas consumidas.[39]
Segundo, es necesario construir una base de datos de referencia con todos los ADN barcodes que probablemente se encuentren en un estudio. Idealmente, estos barcodes deben generarse a partir de especímenes con comprobante depositados en un lugar de acceso público, como un museo de historia natural u otro instituto de investigación.[39] Actualmente, la construcción de estas bases de datos de referencia se está realizando en todo el mundo. Las organizaciones asociadas colaboran en proyectos internacionales como el International Barcode of Life Project (iBOL) y el Consortium for the Barcode of Life (CBOL), que tienen como objetivo construir una referencia de ADN barcode que sirva como base para la identificación de los biomas del mundo. Repositorios de barcode bien conocidos son GenBank del NCBI y el Barcode of Life Data System (BOLD)[39]
Tercero, las células que contienen el ADN de interés deben romperse para exponer su ADN. Este paso, que incluye la extracción y purificación de ADN, debe realizarse en el sustrato bajo investigación. Existen varios procedimientos disponibles para esto.[40] Deben elegirse técnicas específicas para aislar ADN de sustratos con ADN parcialmente degradado, como muestras fósiles y muestras que contienen inhibidores, como sangre, heces y suelo. Las extracciones en las que se espera un bajo rendimiento o calidad de ADN deben realizarse en una instalación de ADN antiguo, junto con protocolos establecidos para evitar la contaminación con ADN moderno.[41][42] Los experimentos deben realizarse siempre por duplicado[43] y con controles positivos incluidos.[39]
Cuarto, deben generarse amplicones a partir del ADN extraído, ya sea de un solo espécimen o de mezclas complejas, utilizando primers basados en los ADN barcodes seleccionados en el paso 1. Para realizar un seguimiento de su origen, deben añadirse nucleótidos etiquetados (ID moleculares o etiquetas MID) en el caso del metabarcoding. Estas etiquetas son necesarias posteriormente en los análisis para rastrear las lecturas de un conjunto de datos masivo hasta su origen.[39]
Quinto, deben seleccionarse las técnicas adecuadas para la secuenciación de ADN. El método clásico de terminación en cadena de Sanger se basa en la incorporación selectiva de inhibidores de elongación de cadena de la ADN polimerasa durante la replicación del ADN. Estas cuatro bases se separan por tamaño mediante electroforesis y luego se identifican mediante detección láser. El método de Sanger es limitado y puede producir una sola lectura a la vez, por lo que es adecuado para generar ADN barcodes de sustratos que contienen solo una especie.[39] Las tecnologías emergentes, como la secuenciación por nanoporos, han reducido el costo de la secuenciación de ADN de unos 30,000 USD por megabyte en 2002 a aproximadamente 0.60 USD en 2016.[44][45] Las tecnologías modernas de secuenciación de nueva generación (NGS) pueden manejar miles o millones de lecturas en paralelo y, por lo tanto, son adecuadas para la identificación masiva de una mezcla de diferentes especies presentes en un sustrato, lo que se resume como metabarcoding[39]
Finalmente, deben realizarse análisis bioinformáticos para comparar los ADN barcodes obtenidos con los Barcode Index Numbers (BIN) en las bibliotecas de referencia.[46] Cada BIN, o clúster BIN, puede identificarse a nivel de especie cuando muestra una alta concordancia (>97%) con los ADN barcodes vinculados a una especie presente en una biblioteca de referencia, o cuando falta la identificación taxonómica a nivel de especie, como una unidad taxonómica operativa (OTU), que se refiere a un grupo de especies (es decir, género, familia o rango taxonómico superior).[39] (Ver agrupación (metagenómica)).Los resultados de la tubería bioinformática deben depurarse, por ejemplo, filtrando lecturas poco fiables, duplicados innecesarios, lecturas de baja calidad y/o lecturas quiméricas. Esto generalmente se realiza mediante búsquedas BLAST en serie en combinación con scripts de filtrado y recorte automáticos.[47] Se necesitan umbrales estandarizados para discriminar entre diferentes especies o entre una identificación correcta e incorrecta[39]
Flujo de trabajo de metabarcoding
A pesar del evidente poder del enfoque, el eDNA metabarcoding se ve afectado por desafíos de precisión y exactitud a lo largo del flujo de trabajo, tanto en el campo, en el laboratorio, como en el análisis computacional.[48] Como se muestra en el diagrama a la derecha, tras el diseño inicial del estudio (hipótesis/pregunta, grupo taxonómico objetivo, etc.), el flujo de trabajo actual de eDNA se compone de tres componentes: campo, laboratorio y bioinformática.[21] El componente de campo incluye la recolección de muestras (por ejemplo, agua, sedimento, aire), las cuales se preservan o congelan antes de la extracción de ADN.El componente de laboratorio sigue cuatro pasos básicos: (i) se concentra el ADN (si no se realizó en el campo) y se purifica; (ii) se usa PCR para amplificar un gen o región objetivo; (iii) se incorporan secuencias únicas de nucleótidos llamadas "indexes" (también conocidas como "barcodes") mediante PCR o se ligan a diferentes productos de PCR, creando una "library" que permite agrupar múltiples muestras; y (iv) las librerías agrupadas se secuencian en una máquina de alto rendimiento. El paso final, después del procesamiento de las muestras en el laboratorio, es procesar computacionalmente los archivos de salida del secuenciador utilizando una pipeline bioinformática robusta[21]
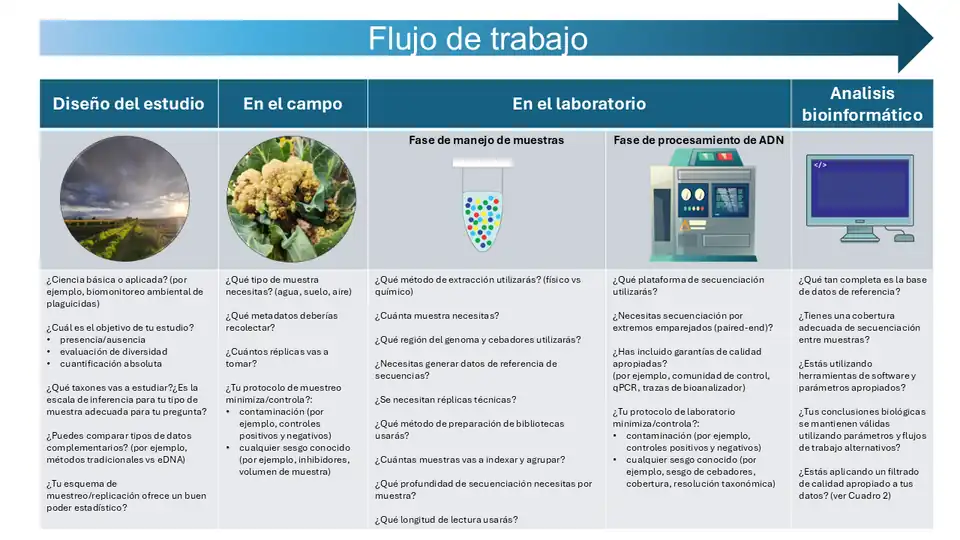
Metodología y visualización
El método requiere que cada ADN recolectado sea archivado con su correspondiente "espécimen tipo" (uno por cada taxón), además de los datos habituales de la colección. Estos tipos se almacenan en instituciones específicas (museos, laboratorios moleculares, universidades, jardines zoológicos, jardines botánicos, herbarios, etc.) uno por cada país, y en algunos casos, se asigna a una misma institución la contención de los tipos de más de un país, en los casos en que algunas naciones no cuentan con la tecnología o los recursos financieros para ello.
De esta manera, la creación de especímenes tipo de códigos genéticos representa una metodología paralela a la llevada a cabo por la taxonomía tradicional.
En una primera etapa, se definió la región del ADN que se utilizaría para realizar el código de barras. Debía ser corta y lograr un alto porcentaje de secuencias únicas. Para animales, algas y hongos, una porción de un gen mitocondrial que codifica para la subunidad 1 de la enzima citocromo oxidasa, CO1, ha proporcionado altos porcentajes (95%), una región en torno a los 648 pares de bases. [49]
En el caso de las plantas, el uso de CO1 no ha sido efectivo ya que presentan bajos niveles de variabilidad en esa región, además de las dificultades que se producen por los frecuentes efectos de poliploidía, introgresión e hibridación, por lo que el genoma del cloroplasto parece más adecuado.[50]
Metabarcoding vs Metagenómica

La metagenómica analiza secuencias del nucleo, cloroplasto o de mitocondrias diversas sin el uso previo de PCR para secuenciar, el metabarcoding se basa en el mismo principio pero se apoya del PCR y utiliza secuencias conservadas, universales y con regiones variables, que pueden servir como código de barras. [51]
Referencias
- ↑ Pierre Taberlet, Aurélie Bonin, Lucie Zinger, Eric Coissac. «cap.8: DNA Metabarcoding data analysis». Environmental DNA: for biodiversity research and monitoring (en inglés). pp. 65-84. Consultado el 14 de noviembre de 2024.
- ↑ a b Hebert, Paul D.N.; Cywinska, Alina; Ball, Shelley L.; deWaard, Jeremy R. (7 de febrero de 2003). «Biological identifications through DNA barcodes». Proceedings of the Royal Society of London. Series B: Biological Sciences (en inglés) 270 (1512): 313-321. ISSN 0962-8452. PMC 1691236. PMID 12614582. doi:10.1098/rspb.2002.2218. Consultado el 14 de noviembre de 2024.
- ↑ Barnes, Matthew A.; Turner, Cameron R. (2016-02). «The ecology of environmental DNA and implications for conservation genetics». Conservation Genetics (en inglés) 17 (1): 1-17. ISSN 1566-0621. doi:10.1007/s10592-015-0775-4. Consultado el 14 de noviembre de 2024.
- ↑ a b c Barnes, Matthew A.; Turner, Cameron R. (1 de febrero de 2016). «The ecology of environmental DNA and implications for conservation genetics». Conservation Genetics (en inglés) 17 (1): 1-17. ISSN 1572-9737. doi:10.1007/s10592-015-0775-4. Consultado el 14 de noviembre de 2024.
- ↑ Deiner, Kristy; Walser, Jean-Claude; Mächler, Elvira; Altermatt, Florian (1 de marzo de 2015). «Choice of capture and extraction methods affect detection of freshwater biodiversity from environmental DNA». Biological Conservation. Special Issue: Environmental DNA: A powerful new tool for biological conservation 183: 53-63. ISSN 0006-3207. doi:10.1016/j.biocon.2014.11.018. Consultado el 14 de noviembre de 2024.
- ↑ Taberlet, Pierre; Coissac, Eric; Pompanon, FrançOis; Brochmann, Christian; Willerslev, Eske (2012-04). «Towards next‐generation biodiversity assessment using DNA metabarcoding». Molecular Ecology (en inglés) 21 (8): 2045-2050. ISSN 0962-1083. doi:10.1111/j.1365-294X.2012.05470.x. Consultado el 14 de noviembre de 2024.
- ↑ a b Bohmann, Kristine; Evans, Alice; Gilbert, M. Thomas P.; Carvalho, Gary R.; Creer, Simon; Knapp, Michael; Yu, Douglas W.; de Bruyn, Mark (2014-06). «Environmental DNA for wildlife biology and biodiversity monitoring». Trends in Ecology & Evolution 29 (6): 358-367. ISSN 0169-5347. doi:10.1016/j.tree.2014.04.003. Consultado el 14 de noviembre de 2024.
- ↑ Goldberg, Caren S.; Turner, Cameron R.; Deiner, Kristy; Klymus, Katy E.; Thomsen, Philip Francis; Murphy, Melanie A.; Spear, Stephen F.; McKee, Anna et al. (2016-11). «Critical considerations for the application of environmental DNA methods to detect aquatic species». En Gilbert, M., ed. Methods in Ecology and Evolution (en inglés) 7 (11): 1299-1307. ISSN 2041-210X. doi:10.1111/2041-210X.12595. Consultado el 14 de noviembre de 2024.
- ↑ Hering, Daniel; Borja, Angel; Jones, J. Iwan; Pont, Didier; Boets, Pieter; Bouchez, Agnes; Bruce, Kat; Drakare, Stina et al. (1 de julio de 2018). «Implementation options for DNA-based identification into ecological status assessment under the European Water Framework Directive». Water Research 138: 192-205. ISSN 0043-1354. doi:10.1016/j.watres.2018.03.003. Consultado el 14 de noviembre de 2024.
- ↑ Ruppert, Krista M.; Kline, Richard J.; Rahman, Md Saydur (1 de enero de 2019). «Past, present, and future perspectives of environmental DNA (eDNA) metabarcoding: A systematic review in methods, monitoring, and applications of global eDNA». Global Ecology and Conservation 17: e00547. ISSN 2351-9894. doi:10.1016/j.gecco.2019.e00547. Consultado el 14 de noviembre de 2024.
- ↑ Ficetola, Gentile Francesco; Taberlet, Pierre; Coissac, Eric (2016-05). «How to limit false positives in environmental DNA and metabarcoding?». Molecular Ecology Resources (en inglés) 16 (3): 604-607. ISSN 1755-098X. doi:10.1111/1755-0998.12508. Consultado el 14 de noviembre de 2024.
- ↑ a b c Creer, Simon; Deiner, Kristy; Frey, Serita; Porazinska, Dorota; Taberlet, Pierre; Thomas, W. Kelley; Potter, Caitlin; Bik, Holly M. (2016-09). «The ecologist's field guide to sequence‐based identification of biodiversity». En Freckleton, Robert, ed. Methods in Ecology and Evolution (en inglés) 7 (9): 1008-1018. ISSN 2041-210X. doi:10.1111/2041-210X.12574. Consultado el 14 de noviembre de 2024.
- ↑ Hering, Daniel; Borja, Angel; Jones, J. Iwan; Pont, Didier; Boets, Pieter; Bouchez, Agnes; Bruce, Kat; Drakare, Stina et al. (1 de julio de 2018). «Implementation options for DNA-based identification into ecological status assessment under the European Water Framework Directive». Water Research 138: 192-205. ISSN 0043-1354. doi:10.1016/j.watres.2018.03.003. Consultado el 14 de noviembre de 2024.
- ↑ Coissac, Eric; Riaz, Tiayyba; Puillandre, Nicolas (2012-04). «Bioinformatic challenges for DNA metabarcoding of plants and animals». Molecular Ecology (en inglés) 21 (8): 1834-1847. ISSN 0962-1083. doi:10.1111/j.1365-294X.2012.05550.x. Consultado el 14 de noviembre de 2024.
- ↑ Yu, Douglas W.; Ji, Yinqiu; Emerson, Brent C.; Wang, Xiaoyang; Ye, Chengxi; Yang, Chunyan; Ding, Zhaoli (2012-08). «Biodiversity soup: metabarcoding of arthropods for rapid biodiversity assessment and biomonitoring». Methods in Ecology and Evolution (en inglés) 3 (4): 613-623. ISSN 2041-210X. doi:10.1111/j.2041-210X.2012.00198.x. Consultado el 14 de noviembre de 2024.
- ↑ Cristescu, Melania E. (2014-10). «From barcoding single individuals to metabarcoding biological communities: towards an integrative approach to the study of global biodiversity». Trends in Ecology & Evolution 29 (10): 566-571. ISSN 0169-5347. doi:10.1016/j.tree.2014.08.001. Consultado el 14 de noviembre de 2024.
- ↑ Gibson, Joel F.; Shokralla, Shadi; Curry, Colin; Baird, Donald J.; Monk, Wendy A.; King, Ian; Hajibabaei, Mehrdad (21 de octubre de 2015). «Large-Scale Biomonitoring of Remote and Threatened Ecosystems via High-Throughput Sequencing». PLOS ONE (en inglés) 10 (10): e0138432. ISSN 1932-6203. PMC 4619546. PMID 26488407. doi:10.1371/journal.pone.0138432. Consultado el 14 de noviembre de 2024.
- ↑ Hering, Daniel; Borja, Angel; Jones, J. Iwan; Pont, Didier; Boets, Pieter; Bouchez, Agnes; Bruce, Kat; Drakare, Stina et al. (1 de julio de 2018). «Implementation options for DNA-based identification into ecological status assessment under the European Water Framework Directive». Water Research 138: 192-205. ISSN 0043-1354. doi:10.1016/j.watres.2018.03.003. Consultado el 14 de noviembre de 2024.
- ↑ a b Ruppert, Krista M.; Kline, Richard J.; Rahman, Md Saydur (1 de enero de 2019). «Past, present, and future perspectives of environmental DNA (eDNA) metabarcoding: A systematic review in methods, monitoring, and applications of global eDNA». Global Ecology and Conservation 17: e00547. ISSN 2351-9894. doi:10.1016/j.gecco.2019.e00547. Consultado el 14 de noviembre de 2024.
- ↑ Margulies, Marcel; Egholm, Michael; Altman, William E.; Attiya, Said; Bader, Joel S.; Bemben, Lisa A.; Berka, Jan; Braverman, Michael S. et al. (2005-09). «Genome sequencing in microfabricated high-density picolitre reactors». Nature (en inglés) 437 (7057): 376-380. ISSN 1476-4687. PMC 1464427. PMID 16056220. doi:10.1038/nature03959. Consultado el 14 de noviembre de 2024.
- ↑ a b c Deiner, Kristy; Bik, Holly M.; Mächler, Elvira; Seymour, Mathew; Lacoursière‐Roussel, Anaïs; Altermatt, Florian; Creer, Simon; Bista, Iliana et al. (2017-11). «Environmental DNA metabarcoding: Transforming how we survey animal and plant communities». Molecular Ecology (en inglés) 26 (21): 5872-5895. ISSN 0962-1083. doi:10.1111/mec.14350. Consultado el 14 de noviembre de 2024.
- ↑ a b c d Deiner, Kristy; Bik, Holly M.; Mächler, Elvira; Seymour, Mathew; Lacoursière‐Roussel, Anaïs; Altermatt, Florian; Creer, Simon; Bista, Iliana et al. (2017-11). «Environmental DNA metabarcoding: Transforming how we survey animal and plant communities». Molecular Ecology (en inglés) 26 (21): 5872-5895. ISSN 0962-1083. doi:10.1111/mec.14350. Consultado el 14 de noviembre de 2024.
- ↑ academic.oup.com. doi:10.1093/nar/gks1195 https://academic.oup.com/nar/article/41/D1/D36/1068219/GenBank
|url=sin título (ayuda). Consultado el 14 de noviembre de 2024. - ↑ Pruesse, E.; Quast, C.; Knittel, K.; Fuchs, B. M.; Ludwig, W.; Peplies, J.; Glockner, F. O. (14 de noviembre de 2007). «SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB». Nucleic Acids Research (en inglés) 35 (21): 7188-7196. ISSN 0305-1048. PMC 2175337. PMID 17947321. doi:10.1093/nar/gkm864. Consultado el 14 de noviembre de 2024.
- ↑ O’Donnell, James L.; Kelly, Ryan P.; Lowell, Natalie C.; Port, Jesse A. (7 de marzo de 2016). «Indexed PCR Primers Induce Template-Specific Bias in Large-Scale DNA Sequencing Studies». PLOS ONE (en inglés) 11 (3): e0148698. ISSN 1932-6203. PMC 4780811. PMID 26950069. doi:10.1371/journal.pone.0148698. Consultado el 14 de noviembre de 2024.
- ↑ SøNstebø, J. H.; Gielly, L.; Brysting, A. K.; Elven, R.; Edwards, M.; Haile, J.; Willerslev, E.; Coissac, E. et al. (2010-11). «Using next‐generation sequencing for molecular reconstruction of past Arctic vegetation and climate». Molecular Ecology Resources (en inglés) 10 (6): 1009-1018. ISSN 1755-098X. doi:10.1111/j.1755-0998.2010.02855.x. Consultado el 14 de noviembre de 2024.
- ↑ Willerslev, Eske; Davison, John; Moora, Mari; Zobel, Martin; Coissac, Eric; Edwards, Mary E.; Lorenzen, Eline D.; Vestergård, Mette et al. (2014-02). «Fifty thousand years of Arctic vegetation and megafaunal diet». Nature (en inglés) 506 (7486): 47-51. ISSN 1476-4687. doi:10.1038/nature12921. Consultado el 14 de noviembre de 2024.
- ↑ Blaxter, Mark; Mann, Jenna; Chapman, Tom; Thomas, Fran; Whitton, Claire; Floyd, Robin; Abebe, Eyualem (29 de octubre de 2005). «Defining operational taxonomic units using DNA barcode data». Philosophical Transactions of the Royal Society B: Biological Sciences (en inglés) 360 (1462): 1935-1943. ISSN 0962-8436. PMC 1609233. PMID 16214751. doi:10.1098/rstb.2005.1725. Consultado el 14 de noviembre de 2024.
- ↑ Deiner, Kristy; Altermatt, Florian (11 de febrero de 2014). «Transport Distance of Invertebrate Environmental DNA in a Natural River». PLOS ONE (en inglés) 9 (2): e88786. ISSN 1932-6203. PMC 3921251. PMID 24523940. doi:10.1371/journal.pone.0088786. Consultado el 14 de noviembre de 2024.
- ↑ Merkes, Christopher M.; McCalla, S. Grace; Jensen, Nathan R.; Gaikowski, Mark P.; Amberg, Jon J. (17 de noviembre de 2014). «Persistence of DNA in Carcasses, Slime and Avian Feces May Affect Interpretation of Environmental DNA Data». PLOS ONE (en inglés) 9 (11): e113346. ISSN 1932-6203. PMC 4234652. PMID 25402206. doi:10.1371/journal.pone.0113346. Consultado el 14 de noviembre de 2024.
- ↑ Yoccoz, N. G.; BråThen, K. A.; Gielly, L.; Haile, J.; Edwards, M. E.; Goslar, T.; Von STEDINGK, H.; Brysting, A. K. et al. (2012-08). «DNA from soil mirrors plant taxonomic and growth form diversity». Molecular Ecology (en inglés) 21 (15): 3647-3655. ISSN 0962-1083. doi:10.1111/j.1365-294X.2012.05545.x. Consultado el 14 de noviembre de 2024.
- ↑ Kress, W. John; García-Robledo, Carlos; Uriarte, Maria; Erickson, David L. (2015-01). «DNA barcodes for ecology, evolution, and conservation». Trends in Ecology & Evolution 30 (1): 25-35. ISSN 0169-5347. doi:10.1016/j.tree.2014.10.008. Consultado el 14 de noviembre de 2024.
- ↑ Galan, Maxime; Pagès, Marie; Cosson, Jean-François (7 de noviembre de 2012). «Next-Generation Sequencing for Rodent Barcoding: Species Identification from Fresh, Degraded and Environmental Samples». PLOS ONE (en inglés) 7 (11): e48374. ISSN 1932-6203. PMC 3492341. PMID 23144869. doi:10.1371/journal.pone.0048374. Consultado el 14 de noviembre de 2024.
- ↑ Ivanova, Natalia V.; Clare, Elizabeth L.; Borisenko, Alex V. (2012). Kress, W. John, ed. DNA Barcoding in Mammals (en inglés). Humana Press. pp. 153-182. ISBN 978-1-61779-591-6. doi:10.1007/978-1-61779-591-6_8. Consultado el 14 de noviembre de 2024.
- ↑ Clare, Elizabeth L. (2014-11). «Molecular detection of trophic interactions: emerging trends, distinct advantages, significant considerations and conservation applications». Evolutionary Applications (en inglés) 7 (9): 1144-1157. ISSN 1752-4571. PMC 4231602. PMID 25553074. doi:10.1111/eva.12225. Consultado el 14 de noviembre de 2024.
- ↑ Orlando, Ludovic; Hänni, Catherine; Douady, Christophe J. (2007-01). «Mammoth and Elephant Phylogenetic Relationships: Mammut Americanum , the Missing Outgroup». Evolutionary Bioinformatics (en inglés) 3. ISSN 1176-9343. doi:10.1177/117693430700300019. Consultado el 14 de noviembre de 2024.
- ↑ Pompanon, Francois; Deagle, Bruce E.; Symondson, William O. C.; Brown, David S.; Jarman, Simon N.; Taberlet, Pierre (2012-04). «Who is eating what: diet assessment using next generation sequencing». Molecular Ecology (en inglés) 21 (8): 1931-1950. ISSN 0962-1083. doi:10.1111/j.1365-294X.2011.05403.x. Consultado el 14 de noviembre de 2024.
- ↑ Deagle, Bruce E.; Jarman, Simon N.; Coissac, Eric; Pompanon, François; Taberlet, Pierre (2014-09). «DNA metabarcoding and the cytochrome c oxidase subunit I marker: not a perfect match». Biology Letters (en inglés) 10 (9): 20140562. ISSN 1744-9561. PMC 4190964. PMID 25209199. doi:10.1098/rsbl.2014.0562. Consultado el 14 de noviembre de 2024.
- ↑ a b c d e f g h i Haarsma, Anne‐Jifke; Siepel, Henk; Gravendeel, Barbara (2016-10). «Added value of metabarcoding combined with microscopy for evolutionary studies of mammals». Zoologica Scripta (en inglés) 45 (S1): 37-49. ISSN 0300-3256. doi:10.1111/zsc.12214. Consultado el 14 de noviembre de 2024.
- ↑ Dhaliwal, Anandika (20 de noviembre de 2022). «DNA Extraction and Purification». Materials and Methods (en inglés). doi:10.13070/mm.en.3.191. Consultado el 14 de noviembre de 2024.
- ↑ Cooper, Alan; Poinar, Hendrik N. (18 de agosto de 2000). «Ancient DNA: Do It Right or Not at All». Science (en inglés) 289 (5482): 1139-1139. ISSN 0036-8075. doi:10.1126/science.289.5482.1139b. Consultado el 14 de noviembre de 2024.
- ↑ Willerslev, Eske; Cooper, Alan (7 de enero de 2005). «Review Paper. Ancient DNA». Proceedings of the Royal Society B: Biological Sciences (en inglés) 272 (1558): 3-16. ISSN 0962-8452. PMC 1634942. PMID 15875564. doi:10.1098/rspb.2004.2813. Consultado el 14 de noviembre de 2024.
- ↑ Ficetola, Gentile F.; Pansu, Johan; Bonin, Aurélie; Coissac, Eric; Giguet‐Covex, Charline; De Barba, Marta; Gielly, Ludovic; Lopes, Carla M. et al. (2015-05). «Replication levels, false presences and the estimation of the presence/absence from eDNA metabarcoding data». Molecular Ecology Resources (en inglés) 15 (3): 543-556. ISSN 1755-098X. doi:10.1111/1755-0998.12338. Consultado el 14 de noviembre de 2024.
- ↑ Shendure, Jay; Aiden, Erez Lieberman (2012-11). «The expanding scope of DNA sequencing». Nature Biotechnology (en inglés) 30 (11): 1084-1094. ISSN 1546-1696. PMC 4149750. PMID 23138308. doi:10.1038/nbt.2421. Consultado el 14 de noviembre de 2024.
- ↑ Panda, Darshan; Molla, Kutubuddin Ali; Baig, Mirza Jainul; Swain, Alaka; Behera, Deeptirekha; Dash, Manaswini (4 de mayo de 2018). «DNA as a digital information storage device: hope or hype?». 3 Biotech (en inglés) 8 (5): 239. ISSN 2190-5738. PMC 5935598. PMID 29744271. doi:10.1007/s13205-018-1246-7. Consultado el 14 de noviembre de 2024.
- ↑ Ratnasingham, Sujeevan; Hebert, Paul D. N. (8 de julio de 2013). «A DNA-Based Registry for All Animal Species: The Barcode Index Number (BIN) System». PLOS ONE (en inglés) 8 (7): e66213. ISSN 1932-6203. PMC 3704603. PMID 23861743. doi:10.1371/journal.pone.0066213. Consultado el 14 de noviembre de 2024.
- ↑ Nelson, Karen E. (3 de enero de 2015). Encyclopedia of Metagenomics: Genes, Genomes and Metagenomes. Basics, Methods, Databases and Tools (en inglés). Springer US. ISBN 978-1-4899-7477-8. Consultado el 14 de noviembre de 2024.
- ↑ Abarenkov, Kessy; Henrik Nilsson, R.; Larsson, Karl‐Henrik; Alexander, Ian J.; Eberhardt, Ursula; Erland, Susanne; Høiland, Klaus; Kjøller, Rasmus et al. (2010-04). «The UNITE database for molecular identification of fungi – recent updates and future perspectives». New Phytologist (en inglés) 186 (2): 281-285. ISSN 0028-646X. doi:10.1111/j.1469-8137.2009.03160.x. Consultado el 14 de noviembre de 2024.
- ↑
- ↑ NEWMASTER, S. G.; FAZEKAS, A. J.; STEEVES, R. A. D.; JANOVEC, J. (2008-05). «Testing candidate plant barcode regions in the Myristicaceae». Molecular Ecology Resources 8 (3): 480-490. ISSN 1755-098X. doi:10.1111/j.1471-8286.2007.02002.x. Consultado el 14 de noviembre de 2024.
- ↑ «Mantenimiento». revistaciencia.uat.edu.mx. doi:10.29059/cienciauat.v16i1.1509. Consultado el 14 de noviembre de 2024.
Enlaces externos
- Esta obra contiene una traducción derivada de «Metabarcoding » de Wikipedia en inglés, concretamente de esta versión, publicada por sus editores bajo la Licencia de documentación libre de GNU y la Licencia Creative Commons Atribución-CompartirIgual 4.0 Internacional.
| Control de autoridades |
|
|---|
 Datos: Q51237016
Datos: Q51237016