Acondrogénesis
| Acondrogénesis | ||
|---|---|---|
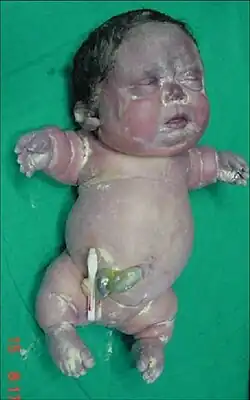 Acondrogénesis tipo I | ||
| Especialidad | genética médica | |
La acondrogénesis es un grupo de enfermedades que se caracterizan por una grave afectación en el desarrollo del hueso y el cartílago, provocando miembros y tronco muy cortos, así como diferentes trastornos del esqueleto. Entran dentro del grupo de enfermedades por trastornos del colágeno tipo II. Son el grupo más grave de las osteocondrodisplasia. Los niños afectados mueren generalmente antes de nacer o poco después del parto. Se distinguen 3 formas de la enfermedad que se denominan 1A, 1B y 2. La frecuencia de aparición es escasa, por lo que se considera una enfermedad rara, presentándose aproximadamente un caso por cada 50.000 nacimientos. Las formas 1A y 1B se heredan según un patrón autosómico recesivo, mientras que la 2 es de tipo autosómico dominante y casi siempre se debe a una mutación nueva por lo que no se hereda de los progenitores.[1]
|
Características radiográficas distintivas
Osificación ausente o severamente retrasada de los cuerpos vertebrales; costillas cortas; osificación ausente de los huesos púbicos, sacro y huesos isquiáticos e ilíacos (pequeños con márgenes internos e inferiores en forma de medialuna); huesos tubulares muy cortos con osificación retrasada en los centros de osificación epifisaria femoral distal y tibial proximal[3]
Referencias
- ↑ Achondrogenesis. Genetics Home Reference. Consultado el 17 de enero de 2013.
- ↑ «GARD». Consultado el 28 de agosto de 2025.
- ↑ Gregersen, Pernille Axél (24 de octubre de 2024). «Table 1. [Clinical and Radiographic Features of Type II Collagen Disorders from Most to Least Severe].». www.ncbi.nlm.nih.gov (en inglés). Consultado el 29 de agosto de 2025.
 Datos: Q2823145
Datos: Q2823145 Multimedia: Achondrogenesis / Q2823145
Multimedia: Achondrogenesis / Q2823145